MSC.sTRAIL Has Better Efficacy than MSC.FL-TRAIL and in Combination with AKTi Blocks Pro-Metastatic Cytokine Production in Prostate Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
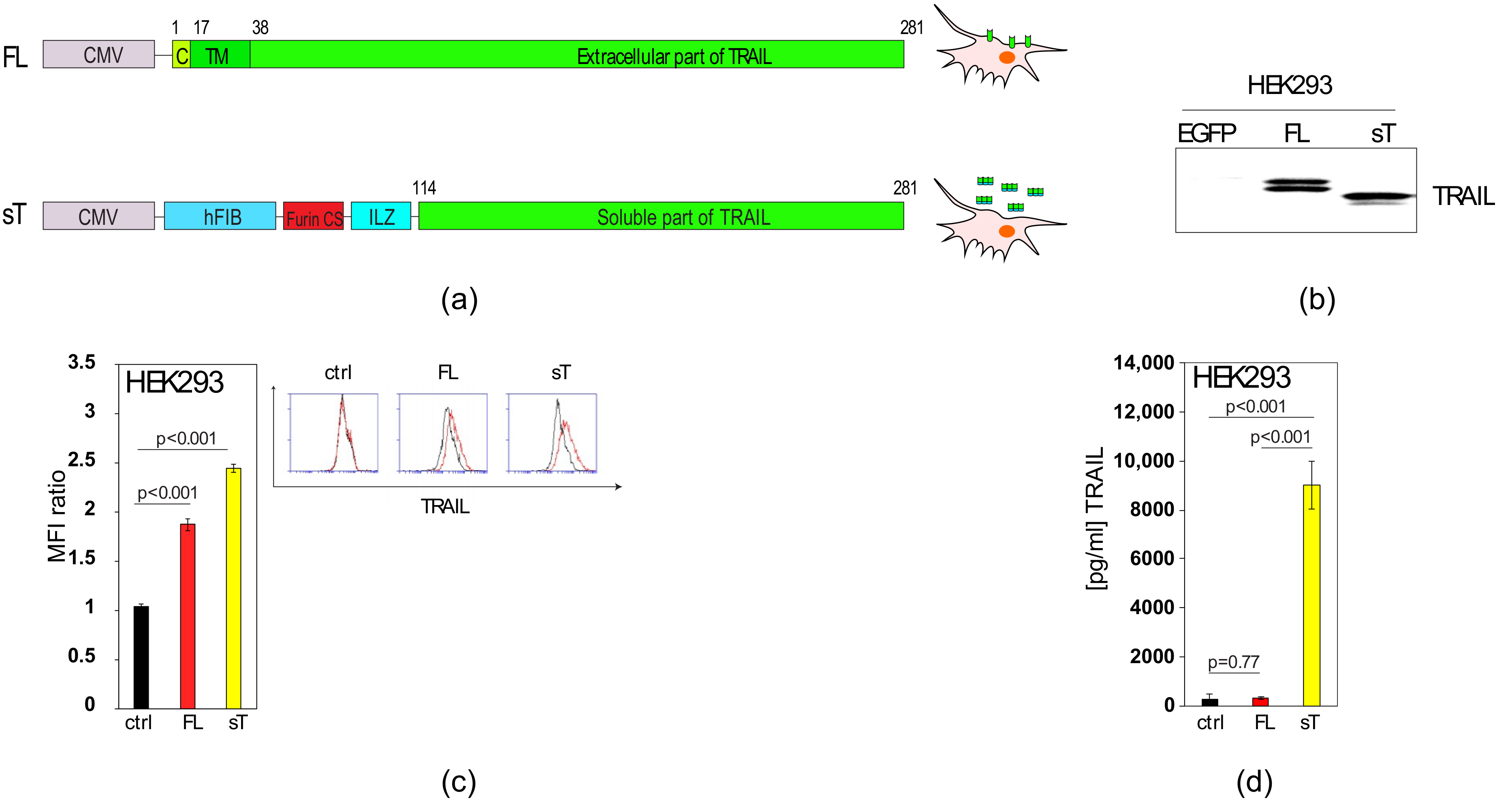
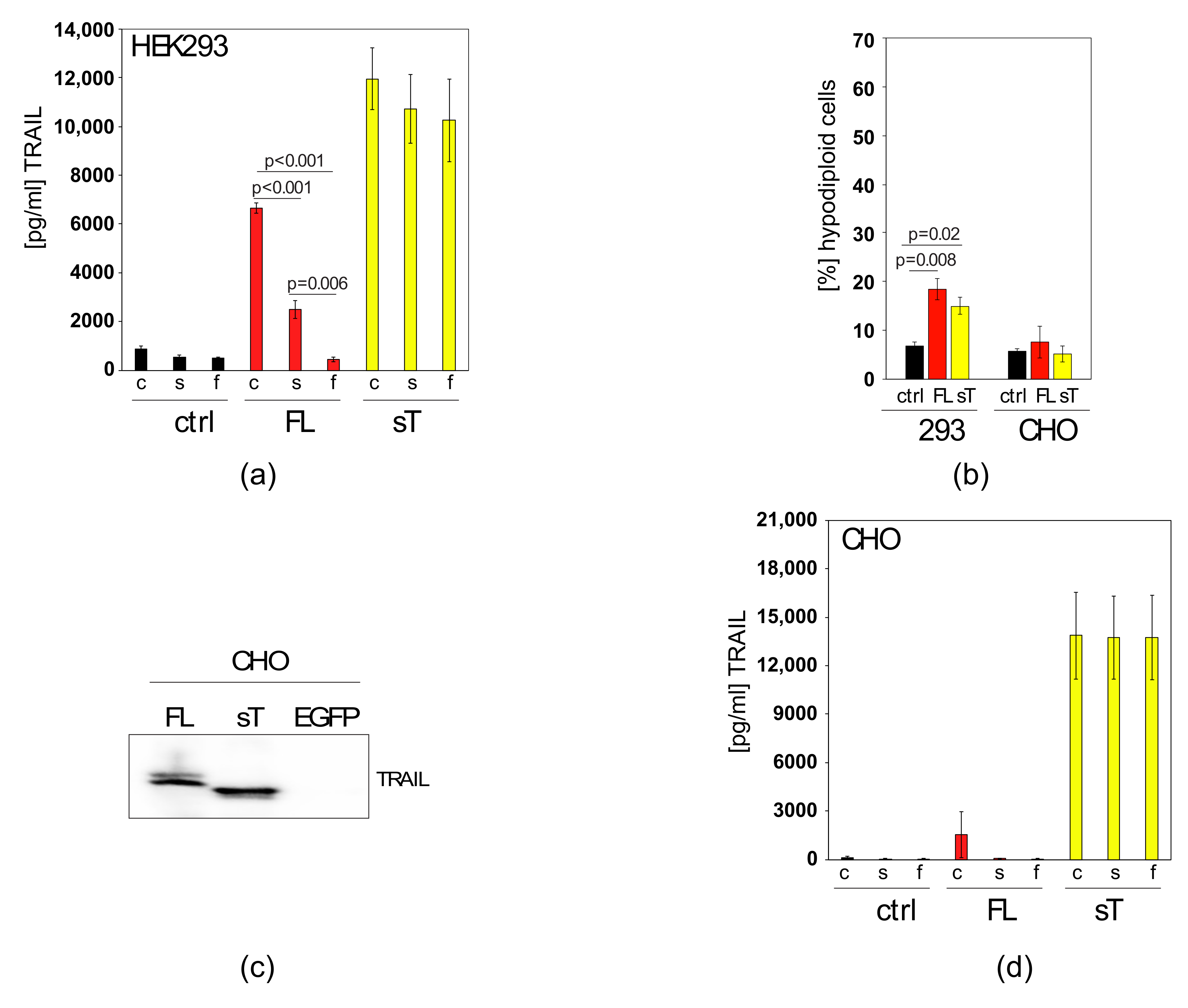
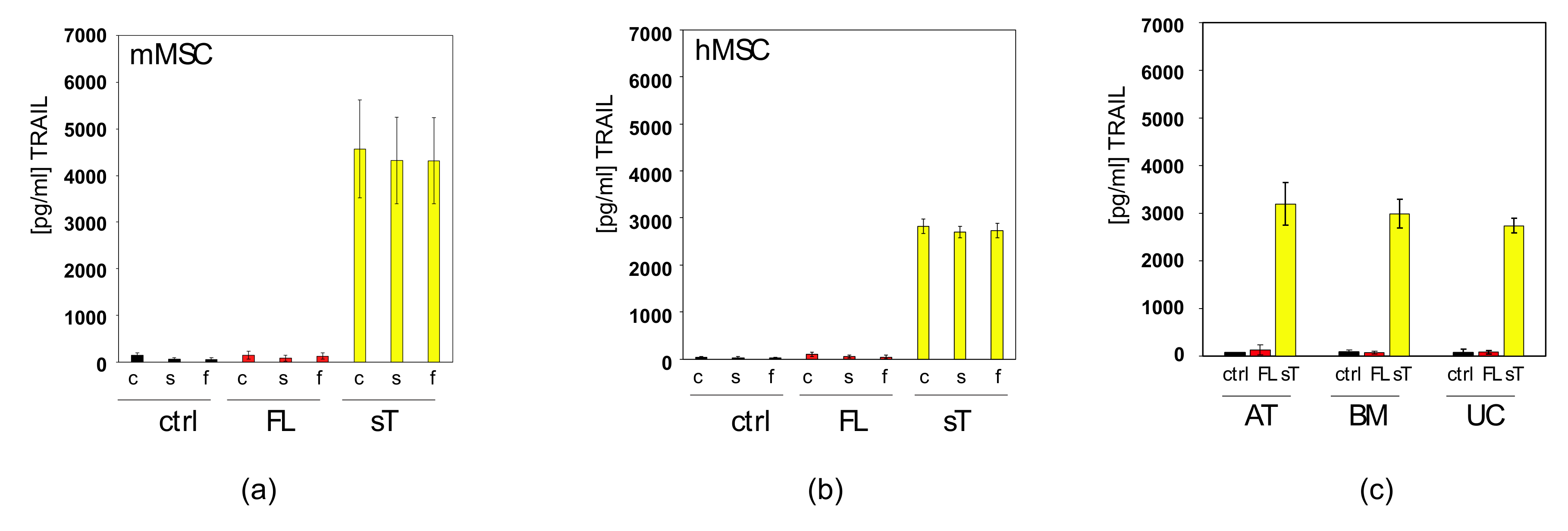
2.1. Comparison of sTRAIL and FL-TRAIL
2.2. Apoptotic Cells Release TRAIL-Containing Cellular Fragments Mimicking Secretion
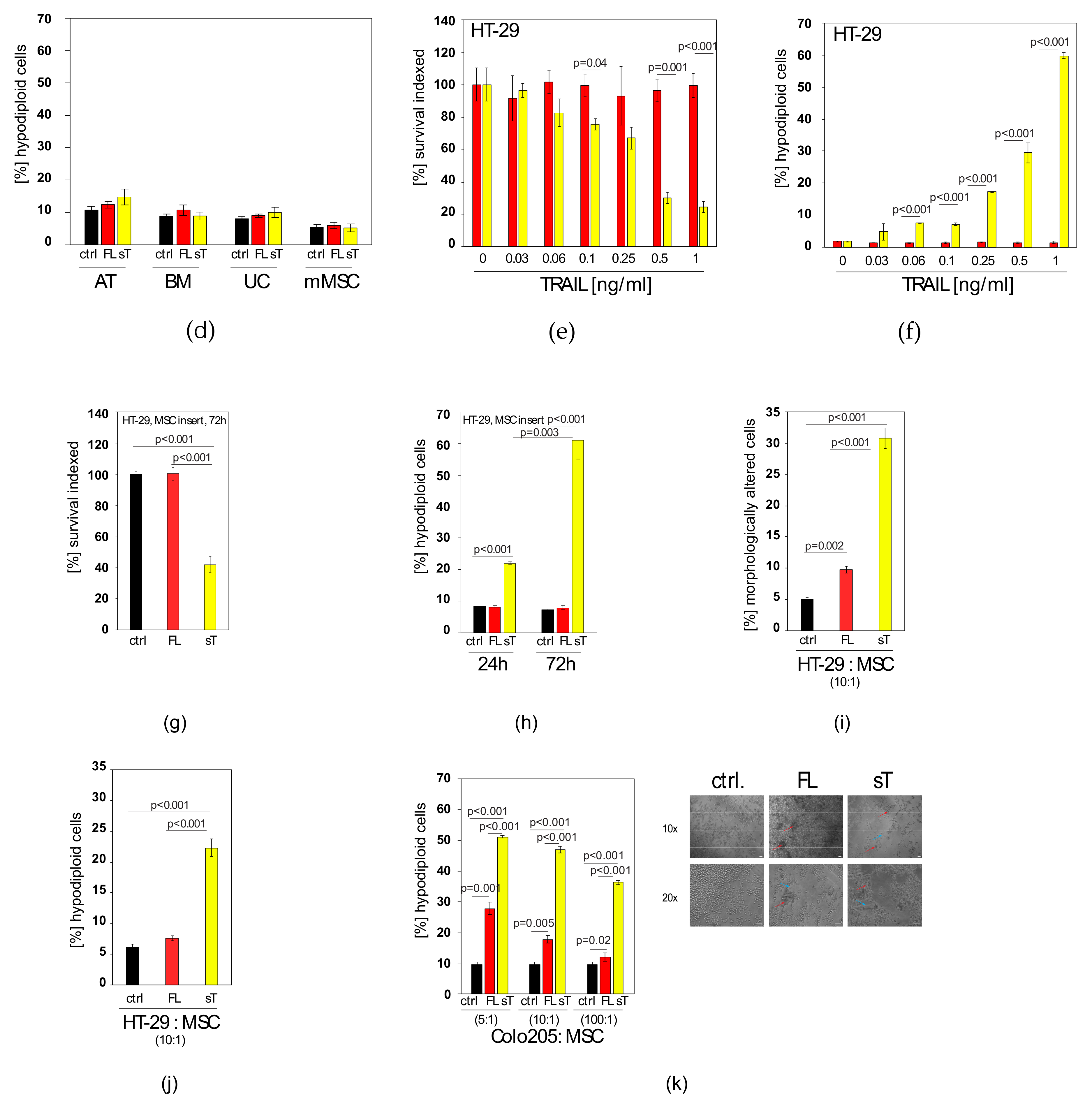
2.3. MSC.sTRAIL Exhibits More Potent Effects on 2D- and 3D-Cultures of Cancer Cells than MSC.FL-TRAIL
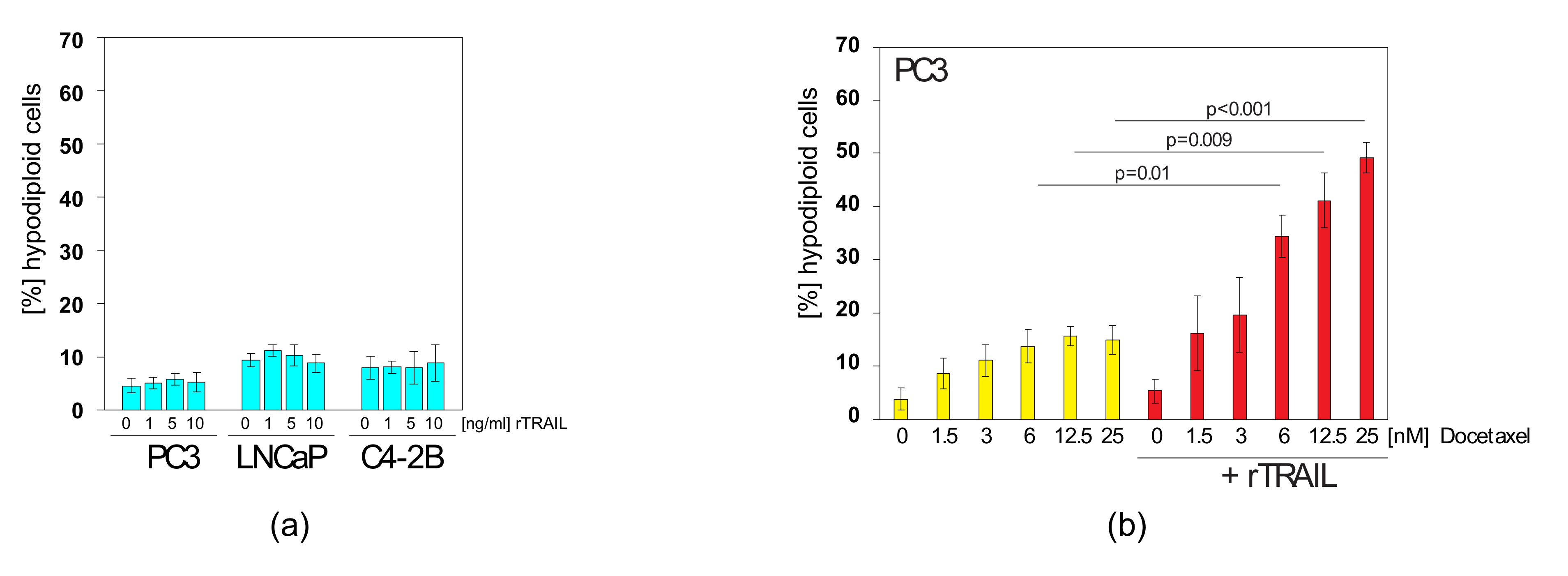
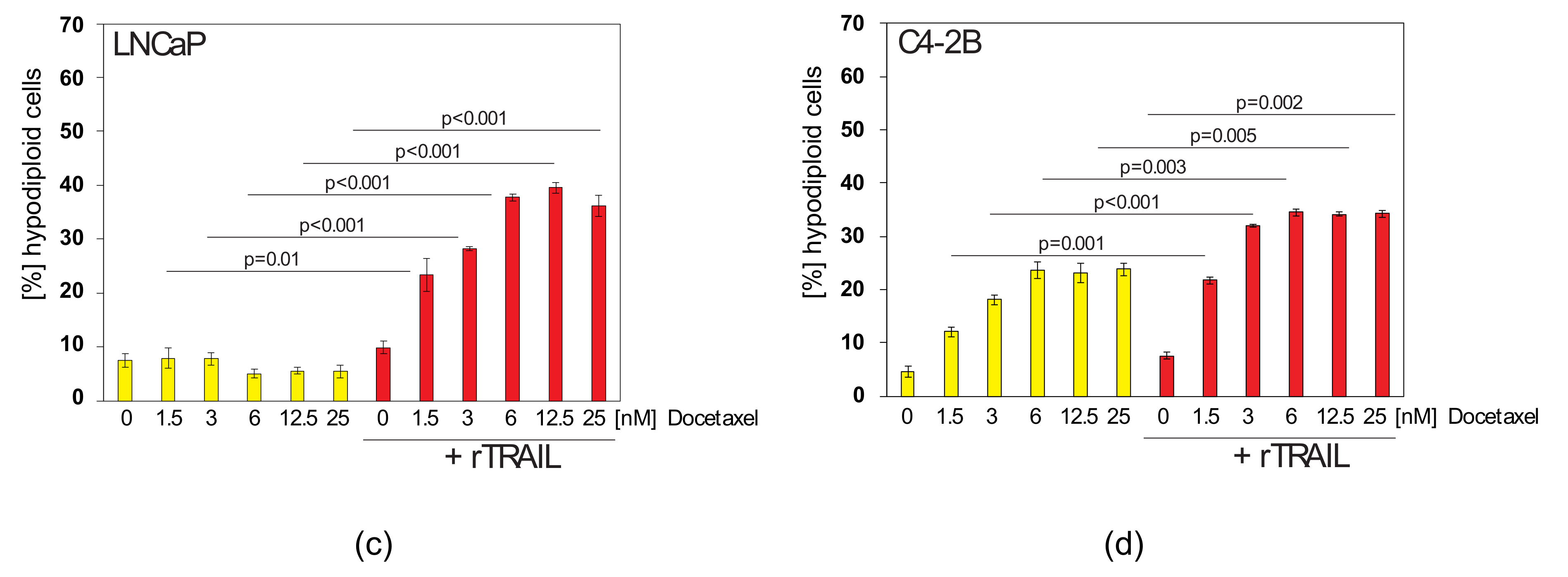
2.4. Docetaxel can Sensitise TRAIL-Resistant Prostate Cancer Cells to TRAIL-Induced Apoptosis
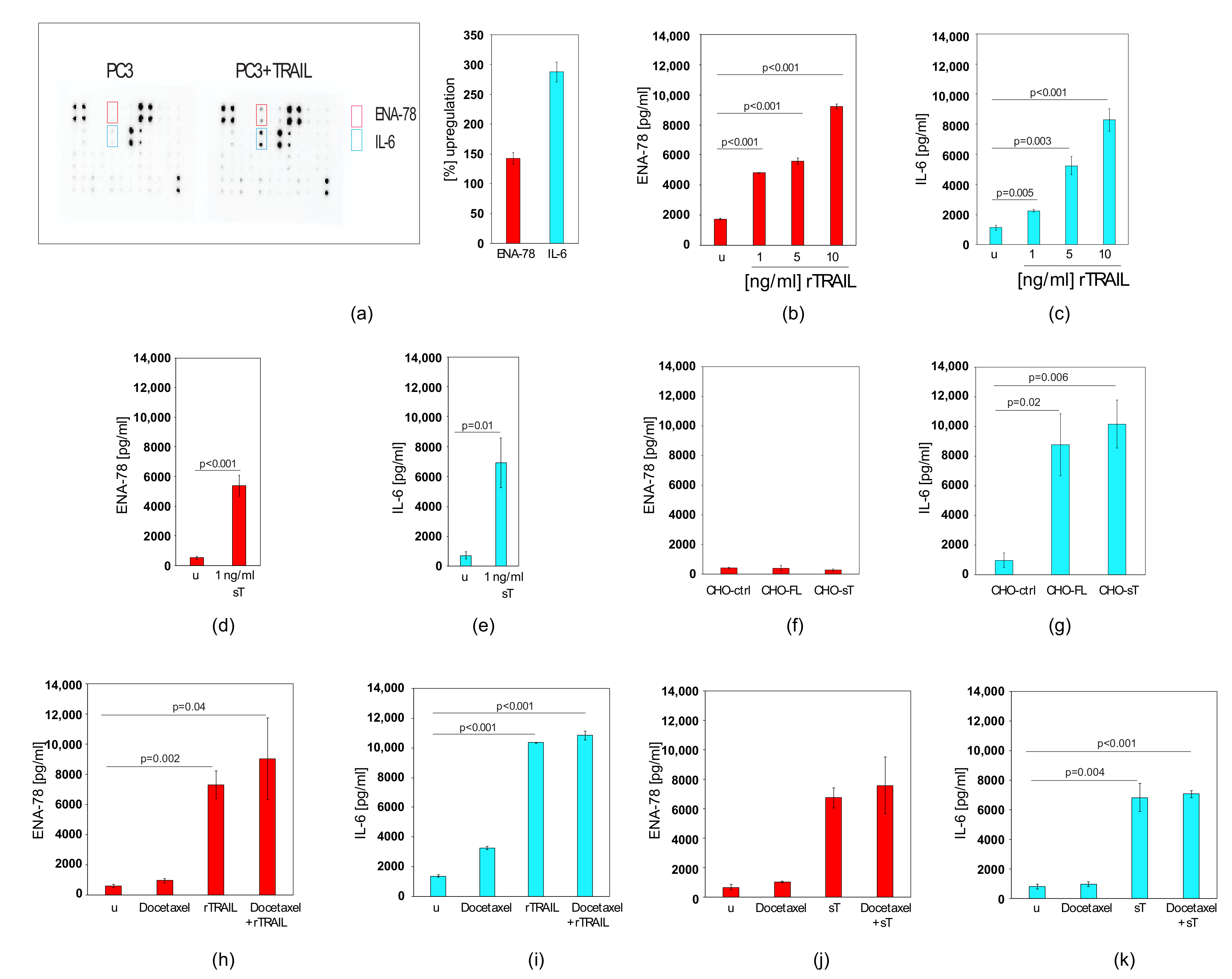
2.5. TRAIL Induces CXCL5/ENA-78 and IL-6 in Prostate Cancer Cells, which cannot be Blocked by Docetaxel Co-treatment
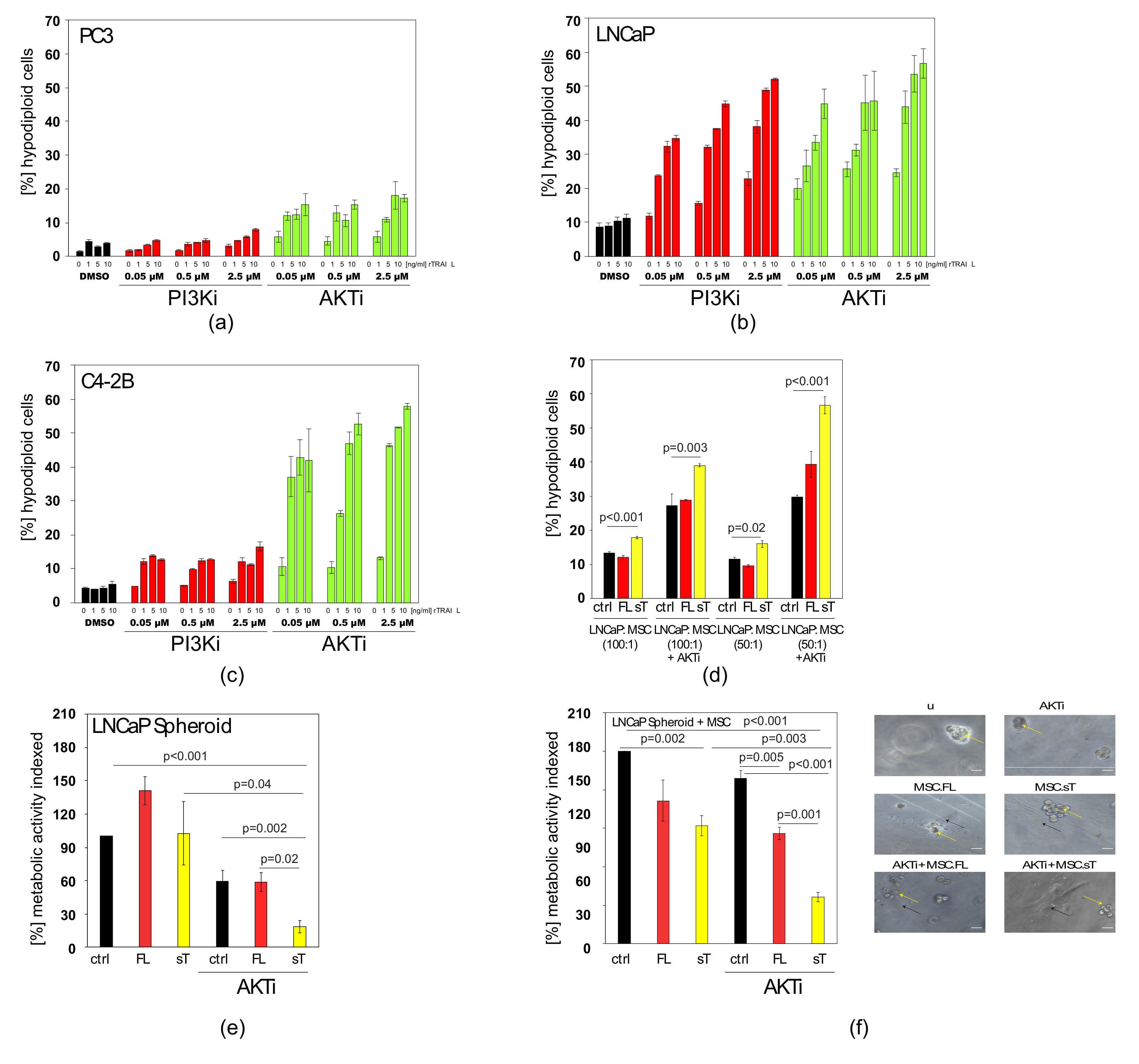
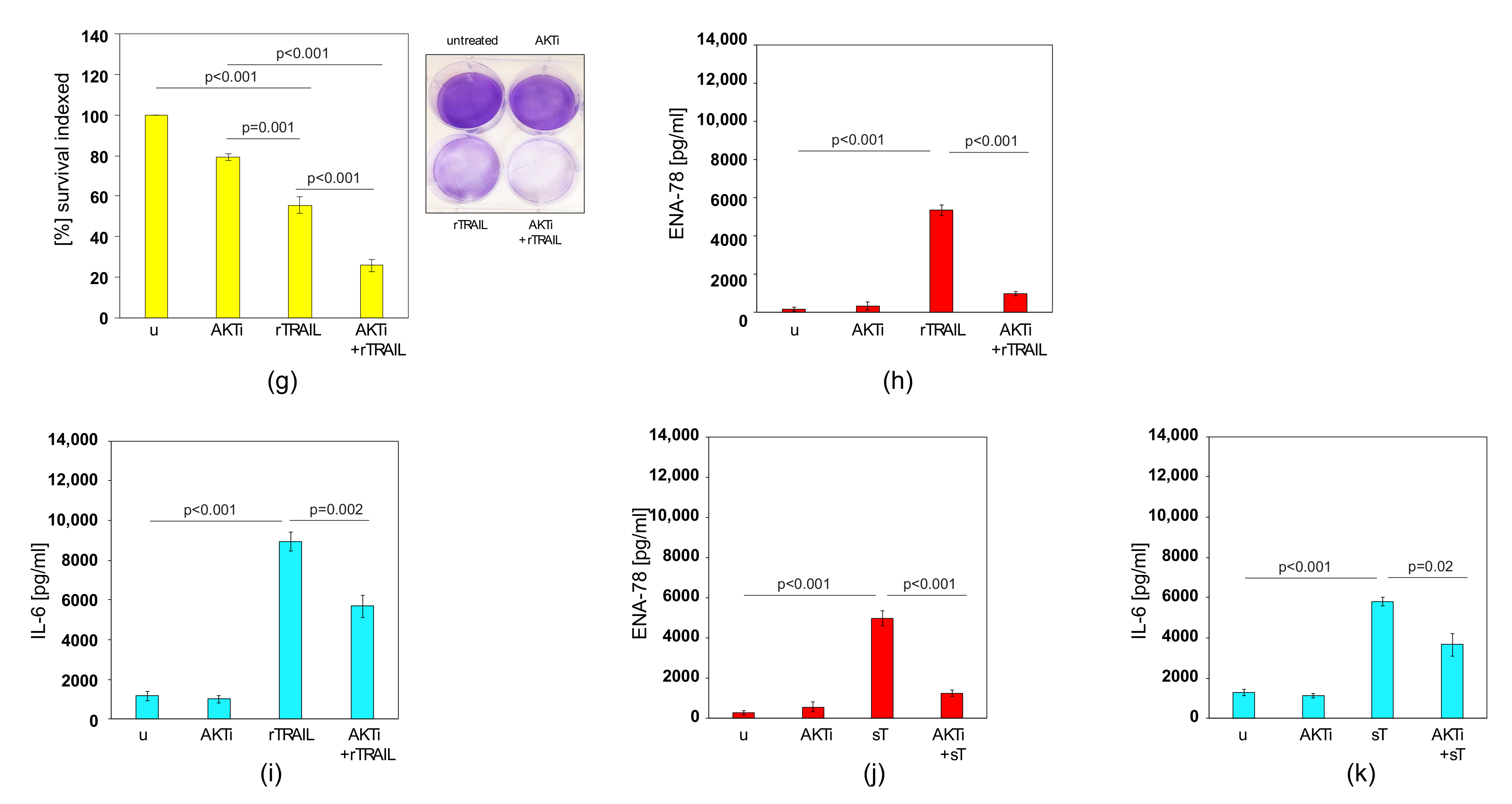
2.6. AKTi can Sensitise Prostate Cancer Cells to TRAIL and Block Cytokine Production
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Generation of TRAIL Constructs
4.3. Transwell co-Cultures
4.4. 3D Spheroid Colorimetric Proliferation/Viability Assay
4.5. 3D Culture
4.6. Crystal Violet Assay
4.7. ELISA
4.8. Cell Surface Staining and FACS Analyses
4.9. Apoptosis Assays
4.10. Western Blot
4.11. Cytokine Array
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duiker, E.W.; Mom, C.H.; de Jong, S.; Willemse, P.H.; Gietema, J.A.; van der Zee, A.G.; de Vries, E.G. The clinical trail of trail. Eur. J. Cancer 2006, 42, 2233–2240. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting trail back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharm. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the tnf family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Wu, G.S. Trail as a target in anti-cancer therapy. Cancer Lett. 2009, 285, 1–5. [Google Scholar] [CrossRef]
- Kretz, A.L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. Trailblazing strategies for cancer treatment. Cancers 2019, 11, 456. [Google Scholar] [CrossRef]
- Nagane, M.; Huang, H.J.; Cavenee, W.K. The potential of trail for cancer chemotherapy. Apoptosis 2001, 6, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the tnfr family, and dr4 induce fadd- dependent apoptosis and activate the nf-kappab pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Mahalingam, D.; Szegezdi, E.; Keane, M.; de Jong, S.; Samali, A. Trail receptor signalling and modulation: Are we on the right trail? Cancer Treat. Rev. 2009, 35, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. Trail receptors 1 (dr4) and 2 (dr5) signal fadd-dependent apoptosis and activate nf-kappab. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef]
- Hellwig, C.T.; Rehm, M. Trail signaling and synergy mechanisms used in trail-based combination therapies. Mol. Cancer 2012, 11, 3–13. [Google Scholar] [CrossRef]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. Fadd/mort1 and caspase-8 are recruited to trail receptors 1 and 2 and are essential for apoptosis mediated by trail receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef]
- Jager, R.; Zwacka, R.M. The enigmatic roles of caspases in tumor development. Cancers 2010, 2, 1952–1979. [Google Scholar] [CrossRef] [PubMed]
- Buneker, C.; Mohr, A.; Zwacka, R.M. The trail-receptor-1: Trail-receptor-3 and -4 ratio is a predictor for trail sensitivity of cancer cells. Oncol. Rep. 2009, 21, 1289–1295. [Google Scholar]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor trail-r4 induces nf-kappab and protects against trail-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F.; Goodwin, R.G.; Smith, C.A. Cloning and characterization of trail-r3, a novel member of the emerging trail receptor family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand trail. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef] [PubMed]
- Naval, J.; de Miguel, D.; Gallego-Lleyda, A.; Anel, A.; Martinez-Lostao, L. Importance of trail molecular anatomy in receptor oligomerization and signaling. Implications for cancer therapy. Cancers 2019, 11, 444. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K. Three is better than one: Pre-ligand receptor assembly in the regulation of tnf receptor signaling. Cytokine 2007, 37, 101–107. [Google Scholar] [CrossRef]
- Clancy, L.; Mruk, K.; Archer, K.; Woelfel, M.; Mongkolsapaya, J.; Screaton, G.; Lenardo, M.J.; Chan, F.K. Preligand assembly domain-mediated ligand-independent association between trail receptor 4 (tr4) and tr2 regulates trail-induced apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 18099–18104. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential inhibition of trail-mediated dr5-disc formation by decoy receptors 1 and 2. Mol. Cell. Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Modulation of trail signaling for cancer therapy. Vitam. Horm. 2004, 67, 275–290. [Google Scholar]
- Fulda, S.; Debatin, K.M. Apoptosis signaling in tumor therapy. Ann. N. Y. Acad. Sci. 2004, 1028, 150–156. [Google Scholar] [CrossRef]
- Edlich, F. Bcl-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates bid, which oligomerizes bak or bax into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic bax and bak: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Exploiting mitochondrial apoptosis for the treatment of cancer. Mitochondrion 2010, 10, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Wick, W.; Weller, M.; Debatin, K.M. Smac agonists sensitize for apo2l/trail- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002, 8, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camidge, D.R.; Herbst, R.S.; Gordon, M.S.; Eckhardt, S.G.; Kurzrock, R.; Durbin, B.; Ing, J.; Tohnya, T.M.; Sager, J.; Ashkenazi, A.; et al. A phase i safety and pharmacokinetic study of the death receptor 5 agonistic antibody pro95780 in patients with advanced malignancies. Clin. Cancer Res. 2010, 16, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.P.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human apo2l/trail) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Trarbach, T.; Moehler, M.; Heinemann, V.; Kohne, C.H.; Przyborek, M.; Schulz, C.; Sneller, V.; Gallant, G.; Kanzler, S. Phase ii trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (trail-r1), in patients with refractory colorectal cancer. Br. J. Cancer 2010, 102, 506–512. [Google Scholar] [CrossRef]
- Fulda, S. Safety and tolerability of trail receptor agonists in cancer treatment. Eur. J. Clin. Pharm. 2015, 71, 525–527. [Google Scholar] [CrossRef]
- Kretz, A.L.; von Karstedt, S.; Hillenbrand, A.; Henne-Bruns, D.; Knippschild, U.; Trauzold, A.; Lemke, J. Should we keep walking along the trail for pancreatic cancer treatment? Revisiting tnf-related apoptosis-inducing ligand for anticancer therapy. Cancers 2018, 10, 77. [Google Scholar] [CrossRef]
- Fakiruddin, K.S.; Ghazalli, N.; Lim, M.N.; Zakaria, Z.; Abdullah, S. Mesenchymal stem cell expressing trail as targeted therapy against sensitised tumour. Int. J. Mol. Sci. 2018, 19, 2188. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Broghammer, E.L. Suppression of tumor growth following intralesional therapy with trail recombinant adenovirus. Mol. Ther. 2001, 4, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Stokes, B.; Kucaba, T.A.; Earel, J.K., Jr.; VanOosten, R.L.; Brincks, E.L.; Norian, L.A. Trail gene therapy: From preclinical development to clinical application. Curr. Gene. 2009, 9, 9–19. [Google Scholar] [CrossRef]
- Mohr, A.; Henderson, G.; Dudus, L.; Herr, I.; Kuerschner, T.; Debatin, K.M.; Weiher, H.; Fisher, K.J.; Zwacka, R.M. Aav-encoded expression of trail in experimental human colorectal cancer leads to tumor regression. Gene Ther. 2004, 11, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Norian, L.A.; James, B.R.; Griffith, T.S. Advances in viral vector-based trail gene therapy for cancer. Cancers 2011, 3, 603–620. [Google Scholar] [CrossRef] [PubMed]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the trails less travelled: Trail in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lim, J.Y.; Park, S.I.; Jeong, C.H.; Oh, J.H.; Jeong, M.; Oh, W.; Park, S.H.; Sung, Y.C.; Jeun, S.S. Gene therapy using trail-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008, 68, 9614–9623. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Eddaoudi, A.; Davies, D.; Janes, S.M. Mesenchymal stem cell delivery of trail can eliminate metastatic cancer. Cancer Res. 2009, 69, 4134–4142. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Lyons, M.; Deedigan, L.; Harte, T.; Shaw, G.; Howard, L.; Barry, F.; O’Brien, T.; Zwacka, R. Mesenchymal stem cells expressing trail lead to tumour growth inhibition in an experimental lung cancer model. J. Cell. Mol. Med. 2008, 12, 2628–2643. [Google Scholar] [CrossRef] [PubMed]
- Spano, C.; Grisendi, G.; Golinelli, G.; Rossignoli, F.; Prapa, M.; Bestagno, M.; Candini, O.; Petrachi, T.; Recchia, A.; Miselli, F.; et al. Soluble trail armed human msc as gene therapy for pancreatic cancer. Sci Rep. 2019, 9, 1788. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Albarenque, S.M.; Deedigan, L.; Yu, R.; Reidy, M.; Fulda, S.; Zwacka, R.M. Targeting of xiap combined with systemic mesenchymal stem cell-mediated delivery of strail ligand inhibits metastatic growth of pancreatic carcinoma cells. Stem Cells 2010, 28, 2109–2120. [Google Scholar] [CrossRef]
- Choi, C.; Kutsch, O.; Park, J.; Zhou, T.; Seol, D.W.; Benveniste, E.N. Tumor necrosis factor-related apoptosis-inducing ligand induces caspase-dependent interleukin-8 expression and apoptosis in human astroglioma cells. Mol. Cell. Biol. 2002, 22, 724–736. [Google Scholar] [CrossRef]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The trail-induced cancer secretome promotes a tumor-supportive immune microenvironment via ccr2. Mol. Cell 2017, 65, 730–742 e735. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Multiple effects of trail in human carcinoma cells: Induction of apoptosis, senescence, proliferation, and cytokine production. Exp. Cell Res. 2008, 314, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, T.; Okamoto, K.; Iwata, J.; Shin, M.; Okamoto, Y.; Yasukochi, A.; Nakayama, K.I.; Kadowaki, T.; Tsukuba, T.; Yamamoto, K. Cathepsin e prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble trail from tumor cell surface. Cancer Res. 2007, 67, 10869–10878. [Google Scholar] [CrossRef]
- Yuan, Z.; Kolluri, K.K.; Sage, E.K.; Gowers, K.H.; Janes, S.M. Mesenchymal stromal cell delivery of full-length tumor necrosis factor-related apoptosis-inducing ligand is superior to soluble type for cancer therapy. Cytotherapy 2015, 17, 885–896. [Google Scholar] [CrossRef]
- Thorburn, A.; Behbakht, K.; Ford, H. Trail receptor-targeted therapeutics: Resistance mechanisms and strategies to avoid them. Drug Resist. Updat. 2008, 11, 17–24. [Google Scholar] [CrossRef]
- Anido-Herranz, U.; Fernandez-Nunez, N.; Afonso-Afonso, J.; Santome-Couto, L.; Medina-Colmenero, A.; Fernandez-Calvo, O.; Lazaro-Quintela, M.; Vazquez, S. Chemotherapy management for unfit patients with metastatic castration-resistant prostate cancer. Clin. Transl. Oncol. 2019, 21, 249–258. [Google Scholar] [CrossRef]
- Eckardt, J.; Eckhardt, G.; Villalona-Calero, M.; Drengler, R.; Von Hoff, D. New anticancer agents in clinical development. Oncology 1995, 9, 1191–1199. [Google Scholar]
- Evdokiou, A.; Bouralexis, S.; Atkins, G.J.; Chai, F.; Hay, S.; Clayer, M.; Findlay, D.M. Chemotherapeutic agents sensitize osteogenic sarcoma cells, but not normal human bone cells, to apo2l/trail-induced apoptosis. Int. J. Cancer 2002, 99, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Deedigan, L.; Albarenque, S.M.; Mohr, A.; Zwacka, R.M. Delivery of strail variants by mscs in combination with cytotoxic drug treatment leads to p53-independent enhanced antitumor effects. Cell Death Dis. 2013, 4, e503. [Google Scholar] [CrossRef]
- Mohr, A.; Deedigan, L.; Jencz, S.; Mehrabadi, Y.; Houlden, L.; Albarenque, S.M.; Zwacka, R.M. Caspase-10: A molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death Differ. 2018, 25, 340–352. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of pten loss in prostate cancer. Nat. Rev. Urol 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. Pten and the pi3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Vlietstra, R.J.; van Alewijk, D.C.; Hermans, K.G.; van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of pten in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar]
- Amanam, I.; Chung, V. Targeted therapies for pancreatic cancer. Cancers 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The erbb/her family of protein-tyrosine kinases and cancer. Pharm. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Rothschild, S.I. Targeted therapies in non-small cell lung cancer-beyond egfr and alk. Cancers 2015, 7, 930–949. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.P.; Luetzkendorf, J.; Widder, M.; Nerger, K.; Caysa, H.; Mueller, T. Trail-transduced multipotent mesenchymal stromal cells (trail-msc) overcome trail resistance in selected crc cell lines in vitro and in vivo. Cancer Gene Ther. 2010, 18, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Studeny, M.; Marini, F.C.; Champlin, R.E.; Zompetta, C.; Fidler, I.J.; Andreeff, M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002, 62, 3603–3608. [Google Scholar] [PubMed]
- Mohr, A.; Zwacka, R. The future of mesenchymal stem cell-based therapeutic approaches for cancer—from cells to ghosts. Cancer Lett. 2018, 414, 239–249. [Google Scholar] [CrossRef]
- Luetzkendorf, J.; Mueller, L.P.; Mueller, T.; Caysa, H.; Nerger, K.; Schmoll, H.J. Growth inhibition of colorectal carcinoma by lentiviral trail-transgenic human mesenchymal stem cells requires their substantial intratumoral presence. J. Cell. Mol. Med. 2010, 14, 2292–2304. [Google Scholar] [CrossRef]
- Menon, L.G.; Kelly, K.; Yang, H.W.; Kim, S.K.; Black, P.M.; Carroll, R.S. Human bone marrow-derived mesenchymal stromal cells expressing s-trail as a cellular delivery vehicle for human glioma therapy. Stem Cells 2009, 27, 2320–2330. [Google Scholar] [CrossRef]
- Moniri, M.R.; Sun, X.Y.; Rayat, J.; Dai, D.; Ao, Z.; He, Z.; Verchere, C.B.; Dai, L.J.; Warnock, G.L. Trail-engineered pancreas-derived mesenchymal stem cells: Characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. 2012, 19, 652–658. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better trails for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.; Gould, S.J. Exosomes and hiv gag bud from endosome-like domains of the t cell plasma membrane. J. Cell Biol. 2006, 172, 923–935. [Google Scholar] [CrossRef]
- Albarenque, S.M.; Zwacka, R.M.; Mohr, A. Both human and mouse mesenchymal stem cells promote breast cancer metastasis. Stem Cell Res. 2011, 7, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Sage, E.K.; Thakrar, R.M.; Janes, S.M. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy 2016, 18, 1435–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Martin, S.J. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory“faddosome” complex upon trail stimulation. Mol. Cell 2017, 65, 715–729 e5. [Google Scholar] [CrossRef]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.R.; Schmid, M.C. Macrophages as key drivers of cancer progression and metastasis. Mediat. Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef]
- Cullen, S.P.; Henry, C.M.; Kearney, C.J.; Logue, S.E.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.C.; Leverkus, M.; Martin, S.J. Fas/cd95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar] [CrossRef]
- Chang, M.S.; McNinch, J.; Basu, R.; Simonet, S. Cloning and characterization of the human neutrophil-activating peptide (ena-78) gene. J. Biol. Chem. 1994, 269, 25277–25282. [Google Scholar]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, M.; Febbraio, M.A.; Whitham, M. From cytokine to myokine: The emerging role of interleukin-6 in metabolic regulation. Immunol. Cell Biol. 2014, 92, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Ohnesorge, N.; Rose-John, S. Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand. J. Immunol. 2006, 63, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Declerck, Y.A. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 2010, 46, 1223–1231. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of stat3 in mediating the cell growth, differentiation and survival signals relayed through the il-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Ishihara, K.; Hirano, T. Il-6 signal transduction and its physiological roles: The signal orchestration model. Rev. Physiol. Biochem. Pharm. 2003, 149, 1–38. [Google Scholar]
- Xie, T.X.; Huang, F.J.; Aldape, K.D.; Kang, S.H.; Liu, M.; Gershenwald, J.E.; Xie, K.; Sawaya, R.; Huang, S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006, 66, 3188–3196. [Google Scholar] [CrossRef]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massague, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahnfeldt, P. Significance of tumor self-seeding as an augmentation to the classic metastasis paradigm. Future Oncol. 2010, 6, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Park, S.S.; Lee, Y.J. Pretreatment of docetaxel enhances trail-mediated apoptosis in prostate cancer cells. J. Cell. Biochem. 2008, 104, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Sellers, W.R. Akt-regulated pathways in prostate cancer. Oncogene 2005, 24, 7465–7474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhou, J.Y.; Wei, W.Z.; Wu, G.S. Activation of the akt survival pathway contributes to trail resistance in cancer cells. PLoS ONE 2010, 5, e10226. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Yu, R.; Zwacka, R.M. Trail-receptor preferences in pancreatic cancer cells revisited: Both trail-r1 and trail-r2 have a licence to kill. BMC Cancer 2015, 15, 494. [Google Scholar] [CrossRef]
- Buneker, C.K.; Yu, R.; Deedigan, L.; Mohr, A.; Zwacka, R.M. Ifn-gamma combined with targeting of xiap leads to increased apoptosis-sensitisation of trail resistant pancreatic carcinoma cells. Cancer Lett. 2012, 316, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Mohr, A.; Zwacka, R.M. In situ trapping of initiator caspases reveals intermediate surprises. Cell Biol. Int. 2007, 31, 526–530. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohr, A.; Chu, T.; Brooke, G.N.; Zwacka, R.M. MSC.sTRAIL Has Better Efficacy than MSC.FL-TRAIL and in Combination with AKTi Blocks Pro-Metastatic Cytokine Production in Prostate Cancer Cells. Cancers 2019, 11, 568. https://doi.org/10.3390/cancers11040568
Mohr A, Chu T, Brooke GN, Zwacka RM. MSC.sTRAIL Has Better Efficacy than MSC.FL-TRAIL and in Combination with AKTi Blocks Pro-Metastatic Cytokine Production in Prostate Cancer Cells. Cancers. 2019; 11(4):568. https://doi.org/10.3390/cancers11040568
Chicago/Turabian StyleMohr, Andrea, Tianyuan Chu, Greg N. Brooke, and Ralf M. Zwacka. 2019. "MSC.sTRAIL Has Better Efficacy than MSC.FL-TRAIL and in Combination with AKTi Blocks Pro-Metastatic Cytokine Production in Prostate Cancer Cells" Cancers 11, no. 4: 568. https://doi.org/10.3390/cancers11040568
APA StyleMohr, A., Chu, T., Brooke, G. N., & Zwacka, R. M. (2019). MSC.sTRAIL Has Better Efficacy than MSC.FL-TRAIL and in Combination with AKTi Blocks Pro-Metastatic Cytokine Production in Prostate Cancer Cells. Cancers, 11(4), 568. https://doi.org/10.3390/cancers11040568