Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TGF-β1/Smad7 Axis in Intestinal Mucosa
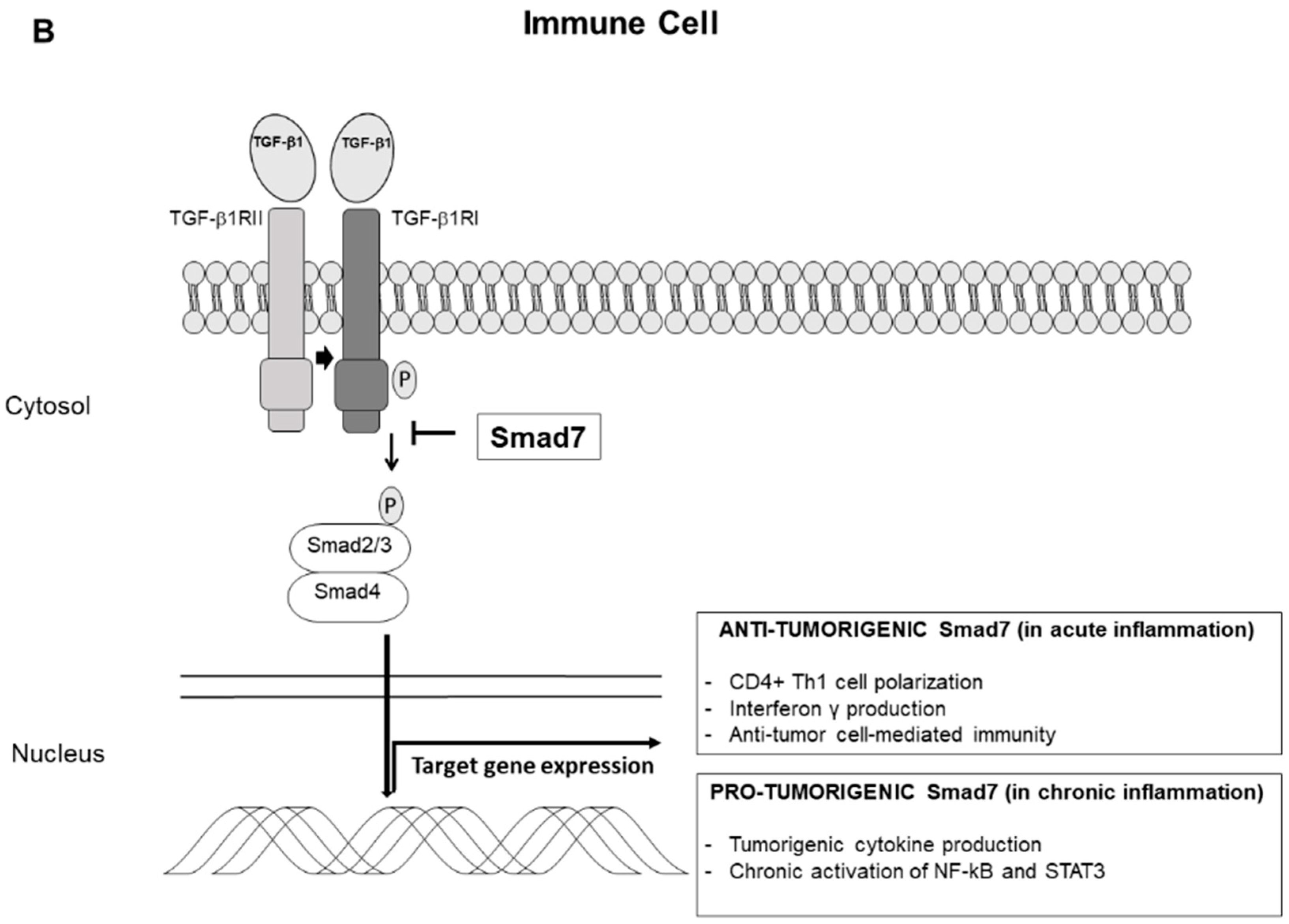
3. Smad7 and Colitis-Associated Colon Cancer
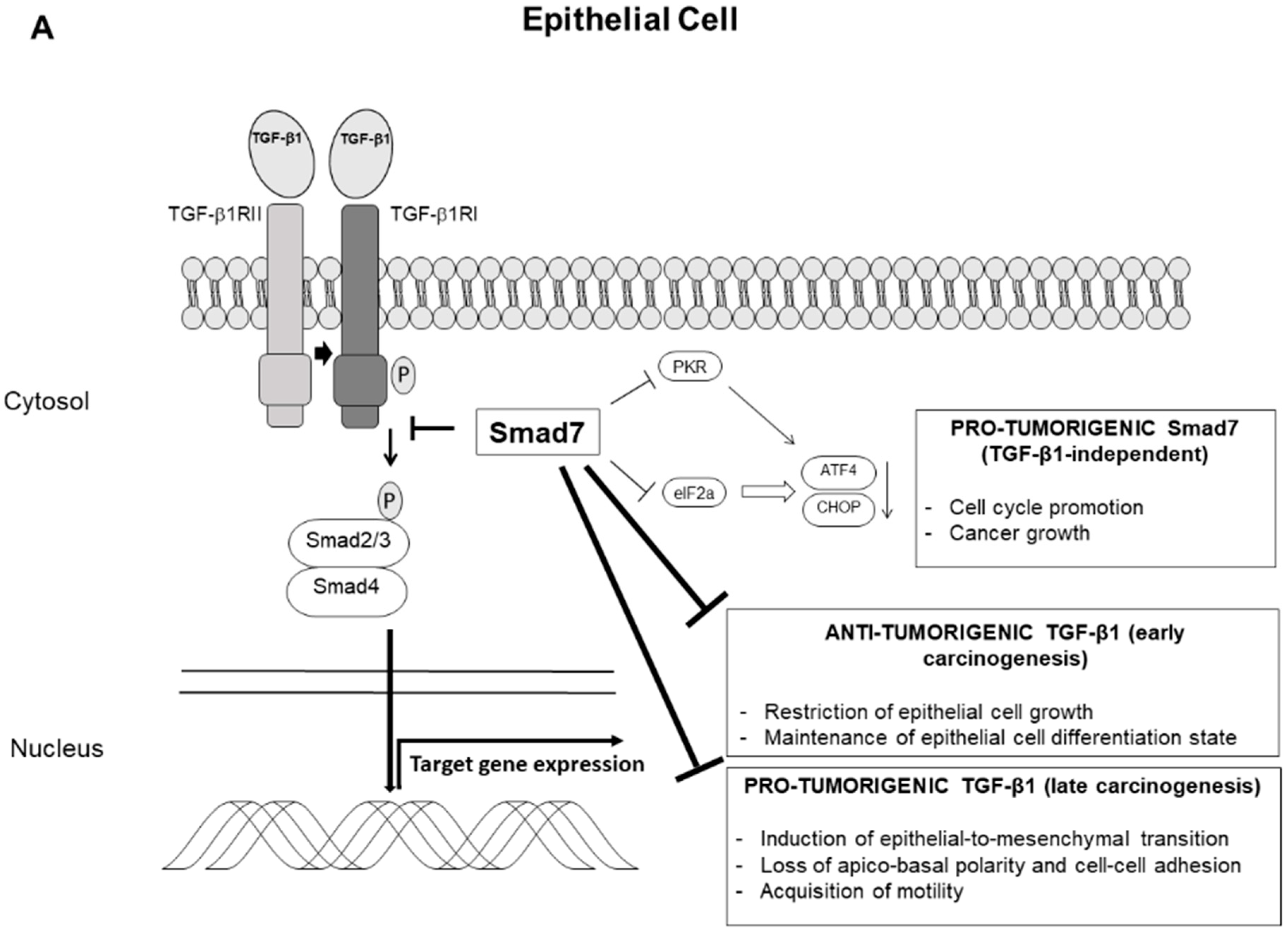
4. Smad7 and Sporadic Colorectal Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Cytokines, IBD, and colitis-associated cancer. Inflamm. Bowel Dis. 2015, 21, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, B.; Peleteiro, B.; Lunet, N. Dietary patterns and colorectal cancer: Systematic review and meta-analysis. Eur. J. Cancer Prev. 2012, 21, 15–23. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P.; Hoops, T.C. The genetic pathogenesis of colorectal cancer. Hematol. Oncol. Clin. N. Am. 2002, 16, 775–810. [Google Scholar] [CrossRef]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2016, 34, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [PubMed]
- Hurwitz, H.I.; Tebbutt, N.C.; Kabbinavar, F.; Giantonio, B.J.; Guan, Z.Z.; Mitchell, L.; Waterkamp, D.; Tabernero, J. Efficacy and safety of bevacizumab in metastatic colorectal cancer: Pooled analysis from seven randomized controlled trials. Oncologist 2013, 18, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Troncone, E.; Marafini, I.; Stolfi, C.; Monteleone, G. Transforming Growth Factor-beta1/Smad7 in Intestinal Immunity, Inflammation, and Cancer. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Shi, W.; Sun, C.; He, B.; Xiong, W.; Shi, X.; Yao, D.; Cao, X. GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J. Cell Biol. 2004, 164, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.H.; Meng, A.; Chen, Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef]
- Salmela, M.T.; Pender, S.L.; Karjalainen-Lindsberg, M.L.; Puolakkainen, P.; Macdonald, T.T.; Saarialho-Kere, U. Collagenase-1 (MMP-1), matrilysin-1 (MMP-7), and stromelysin-2 (MMP-10) are expressed by migrating enterocytes during intestinal wound healing. Scand. J. Gastroenterol. 2004, 39, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Stallmach, A.; Schuppan, D.; Riese, H.H.; Matthes, H.; Riecken, E.O. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology 1992, 102, 1920–1929. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Jackson, C.L.; Pickard, K.M.; Buckley, M.; Rovedatti, L.; Leakey, N.A.; Picariello, L.; Cazzola, P.; Monteleone, G.; Tonelli, F.; et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut 2009, 58, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Gorelik, L.; Fields, P.E.; Flavell, R.A. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J. Immunol. 2000, 165, 4773–4777. [Google Scholar] [CrossRef] [PubMed]
- Heath, V.L.; Murphy, E.E.; Crain, C.; Tomlinson, M.G.; O’Garra, A. TGF-beta1 down-regulates Th2 development and results in decreased IL-4-induced STAT6 activation and GATA-3 expression. Eur. J. Immunol. 2000, 30, 2639–2649. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25-T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Gorham, J.D.; Guler, M.L.; Fenoglio, D.; Gubler, U.; Murphy, K.M. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J. Immunol. 1998, 161, 1664–1670. [Google Scholar]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Thornton, A.M.; Kinney, M.C.; Ma, C.A.; Spinner, J.J.; Fuss, I.J.; Shevach, E.M.; Jain, A. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGF-beta) signaling and the development of regulatory T cells. J. Biol. Chem. 2011, 286, 40520–40530. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Babyatsky, M.W.; Rossiter, G.; Podolsky, D.K. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 1996, 110, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J. Clin. Investig. 2001, 108, 601–609. [Google Scholar] [CrossRef]
- Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stuber, E.; Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 1995, 182, 1281–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boirivant, M.; Fuss, I.J.; Chu, A.; Strober, W. Oxazolone colitis: A murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J. Exp. Med. 1998, 188, 1929–1939. [Google Scholar] [CrossRef]
- Boirivant, M.; Pallone, F.; Di Giacinto, C.; Fina, D.; Monteleone, I.; Marinaro, M.; Caruso, R.; Colantoni, A.; Palmieri, G.; Sanchez, M.; et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology 2006, 131, 1786–1798. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Yakicier, M.C.; Irmak, M.B.; Romano, A.; Kew, M.; Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 1999, 18, 4879–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggins, G.J.; Kinzler, K.W.; Vogelstein, B.; Thiagalingam, S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997, 57, 2578–2580. [Google Scholar] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Katsuno, Y.; Qin, J.; Oses-Prieto, J.; Wang, H.; Jackson-Weaver, O.; Zhang, T.; Lamouille, S.; Wu, J.; Burlingame, A.; Xu, J.; et al. Arginine methylation of SMAD7 by PRMT1 in TGF-beta-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J. Biol. Chem. 2018, 293, 13059–13072. [Google Scholar] [CrossRef]
- De Leeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Rego, R.L.; Ansell, S.M.; Knutson, K.L.; Foster, N.R.; Sargent, D.J. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology 2009, 137, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 1990, 323, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Annese, V.; Daperno, M.; Rutter, M.D.; Amiot, A.; Bossuyt, P.; East, J.; Ferrante, M.; Gotz, M.; Katsanos, K.H.; Kiesslich, R.; et al. European evidence based consensus for endoscopy in inflammatory bowel disease. J. Crohns Colitis 2013, 7, 982–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Wang, K.; Kim, M.K.; Di Caro, G.; Wong, J.; Shalapour, S.; Wan, J.; Zhang, W.; Zhong, Z.; Sanchez-Lopez, E.; Wu, L.W.; et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014, 41, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [PubMed]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Rizzo, A.; Waldner, M.J.; Stolfi, C.; Sarra, M.; Fina, D.; Becker, C.; Neurath, M.F.; Macdonald, T.T.; Pallone, F.; Monteleone, G.; et al. Smad7 expression in T cells prevents colitis-associated cancer. Cancer Res. 2011, 71, 7423–7432. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; De Mare, V.; Rocchi, C.; Stolfi, C.; Colantoni, A.; Neurath, M.F.; Macdonald, T.T.; Pallone, F.; Monteleone, G.; Fantini, M.C. Smad7 induces plasticity in tumor-infiltrating Th17 cells and enables TNF-alpha-mediated killing of colorectal cancer cells. Carcinogenesis 2014, 35, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKbeta acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef]
- Dowdy, S.C.; Mariani, A.; Reinholz, M.M.; Keeney, G.L.; Spelsberg, T.C.; Podratz, K.C.; Janknecht, R. Overexpression of the TGF-beta antagonist Smad7 in endometrial cancer. Gynecol. Oncol. 2005, 96, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Osawa, H.; Nakajima, M.; Kato, H.; Fukuchi, M.; Kuwano, H. Prognostic value of the expression of Smad6 and Smad7, as inhibitory Smads of the TGF-beta superfamily, in esophageal squamous cell carcinoma. Anticancer Res. 2004, 24, 3703–3709. [Google Scholar] [PubMed]
- Leng, A.; Liu, T.; He, Y.; Li, Q.; Zhang, G. Smad4/Smad7 balance: A role of tumorigenesis in gastric cancer. Exp. Mol. Pathol. 2009, 87, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, H.S.; Lee, H.J.; Hur, K.; Kim, W.H.; Bang, Y.J.; Kim, S.J.; Lee, K.U.; Choe, K.J.; Yang, H.K. Prognostic significance of the expression of Smad4 and Smad7 in human gastric carcinomas. Ann. Oncol. 2004, 15, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Cao, T.; Smith, D.A.; Myers, T.E.; Wang, X.J. Smads mediate signaling of the TGFbeta superfamily in normal keratinocytes but are lost during skin chemical carcinogenesis. Oncogene 2001, 20, 471–483. [Google Scholar] [CrossRef]
- Theohari, I.; Giannopoulou, I.; Magkou, C.; Nomikos, A.; Melissaris, S.; Nakopoulou, L. Differential effect of the expression of TGF-beta pathway inhibitors, Smad-7 and Ski, on invasive breast carcinomas: Relation to biologic behavior. APMIS 2012, 120, 92–100. [Google Scholar] [CrossRef]
- Azuma, H.; Ehata, S.; Miyazaki, H.; Watabe, T.; Maruyama, O.; Imamura, T.; Sakamoto, T.; Kiyama, S.; Kiyama, Y.; Ubai, T.; et al. Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC(A) cells. J. Natl. Cancer Inst. 2005, 97, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, C.; Kim, S.J. Smad7 sensitizes tumor necrosis factor induced apoptosis through the inhibition of antiapoptotic gene expression by suppressing activation of the nuclear factor-kappaB pathway. Cancer Res. 2007, 67, 9577–9583. [Google Scholar] [CrossRef]
- Kim, S.; Han, J.; Lee, S.K.; Koo, M.; Cho, D.H.; Bae, S.Y.; Choi, M.Y.; Kim, J.S.; Kim, J.H.; Choe, J.H.; et al. Smad7 acts as a negative regulator of the epidermal growth factor (EGF) signaling pathway in breast cancer cells. Cancer Lett. 2012, 314, 147–154. [Google Scholar] [CrossRef]
- Kaczorowski, M.; Biecek, P.; Donizy, P.; Pieniazek, M.; Matkowski, R.; Halon, A. SMAD7 is a novel independent predictor of survival in patients with cutaneous melanoma. Transl. Res. 2019, 204, 72–81. [Google Scholar] [CrossRef]
- Wang, P.; Fan, J.; Chen, Z.; Meng, Z.Q.; Luo, J.M.; Lin, J.H.; Zhou, Z.H.; Chen, H.; Wang, K.; Xu, Z.D.; et al. Low-level expression of Smad7 correlates with lymph node metastasis and poor prognosis in patients with pancreatic cancer. Ann. Surg. Oncol. 2009, 16, 826–835. [Google Scholar] [CrossRef]
- Arnold, N.B.; Ketterer, K.; Kleeff, J.; Friess, H.; Buchler, M.W.; Korc, M. Thioredoxin is downstream of Smad7 in a pathway that promotes growth and suppresses cisplatin-induced apoptosis in pancreatic cancer. Cancer Res. 2004, 64, 3599–3606. [Google Scholar] [CrossRef]
- Boyer Arnold, N.; Korc, M. Smad7 abrogates transforming growth factor-beta1-mediated growth inhibition in COLO-357 cells through functional inactivation of the retinoblastoma protein. J. Biol. Chem. 2005, 280, 21858–21866. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Ishiwata, T.; Maruyama, H.; Friess, H.; Truong, P.; Buchler, M.W.; Falb, D.; Korc, M. The TGF-beta signaling inhibitor Smad7 enhances tumorigenicity in pancreatic cancer. Oncogene 1999, 18, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Kuang, C.; Xiao, Y.; Liu, X.; Stringfield, T.M.; Zhang, S.; Wang, Z.; Chen, Y. In vivo disruption of TGF-beta signaling by Smad7 leads to premalignant ductal lesions in the pancreas. Proc. Natl. Acad. Sci USA 2006, 103, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Boulay, J.L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. SMAD7 is a prognostic marker in patients with colorectal cancer. Int. J. Cancer 2003, 104, 446–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenesa, A.; Dunlop, M.G. New insights into the etiology of colorectal cancer from genome-wide association studies. Nat. Rev. Genet. 2009, 10, 353–358. [Google Scholar] [CrossRef]
- Phipps, A.I.; Newcomb, P.A.; Garcia-Albeniz, X.; Hutter, C.M.; White, E.; Fuchs, C.S.; Hazra, A.; Ogino, S.; Nan, H.; Ma, J.; et al. Association between colorectal cancer susceptibility loci and survival time after diagnosis with colorectal cancer. Gastroenterology 2012, 143, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, W.; Nie, M.; Li, C.; Wang, L. SMAD7 polymorphisms and colorectal cancer risk: A meta-analysis of case-control studies. Oncotarget 2016, 7, 75561–75570. [Google Scholar] [CrossRef]
- Hu, Y.; Gaedcke, J.; Emons, G.; Beissbarth, T.; Grade, M.; Jo, P.; Yeager, M.; Chanock, S.J.; Wolff, H.; Camps, J.; et al. Colorectal cancer susceptibility loci as predictive markers of rectal cancer prognosis after surgery. Genes Chromosomes Cancer 2018, 57, 140–149. [Google Scholar] [CrossRef]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. Smad7 induces tumorigenicity by blocking TGF-beta-induced growth inhibition and apoptosis. Exp. Cell Res. 2005, 307, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Rachakonda, G.; Deane, N.G.; Datta, P.K. Smad7 induces hepatic metastasis in colorectal cancer. Br. J. Cancer 2008, 99, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zou, C.; Zou, C.; Han, Z.; Xiao, H.; Wei, H.; Wang, W.; Zhang, L.; Zhang, X.; Tang, Q.; et al. MicroRNA-25 functions as a potential tumor suppressor in colon cancer by targeting Smad7. Cancer Lett. 2013, 335, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; De Simone, V.; Colantoni, A.; Franze, E.; Ribichini, E.; Fantini, M.C.; Caruso, R.; Monteleone, I.; Sica, G.S.; Sileri, P.; et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Bevivino, G.; Sedda, S.; Izzo, R.; Laudisi, F.; Dinallo, V.; Franze, E.; Colantoni, A.; Ortenzi, A.; Salvatori, S.; et al. Smad7 knockdown activates protein kinase RNA-associated eIF2alpha pathway leading to colon cancer cell death. Cell Death Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nie, L.; Wu, L.; Liu, Q.; Guo, X. NR2F2 inhibits Smad7 expression and promotes TGF-beta-dependent epithelial-mesenchymal transition of CRC via transactivation of miR-21. Biochem. Biophys. Res. Commun. 2017, 485, 181–188. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troncone, E.; Monteleone, G. Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword. Cancers 2019, 11, 612. https://doi.org/10.3390/cancers11050612
Troncone E, Monteleone G. Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword. Cancers. 2019; 11(5):612. https://doi.org/10.3390/cancers11050612
Chicago/Turabian StyleTroncone, Edoardo, and Giovanni Monteleone. 2019. "Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword" Cancers 11, no. 5: 612. https://doi.org/10.3390/cancers11050612
APA StyleTroncone, E., & Monteleone, G. (2019). Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword. Cancers, 11(5), 612. https://doi.org/10.3390/cancers11050612