Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
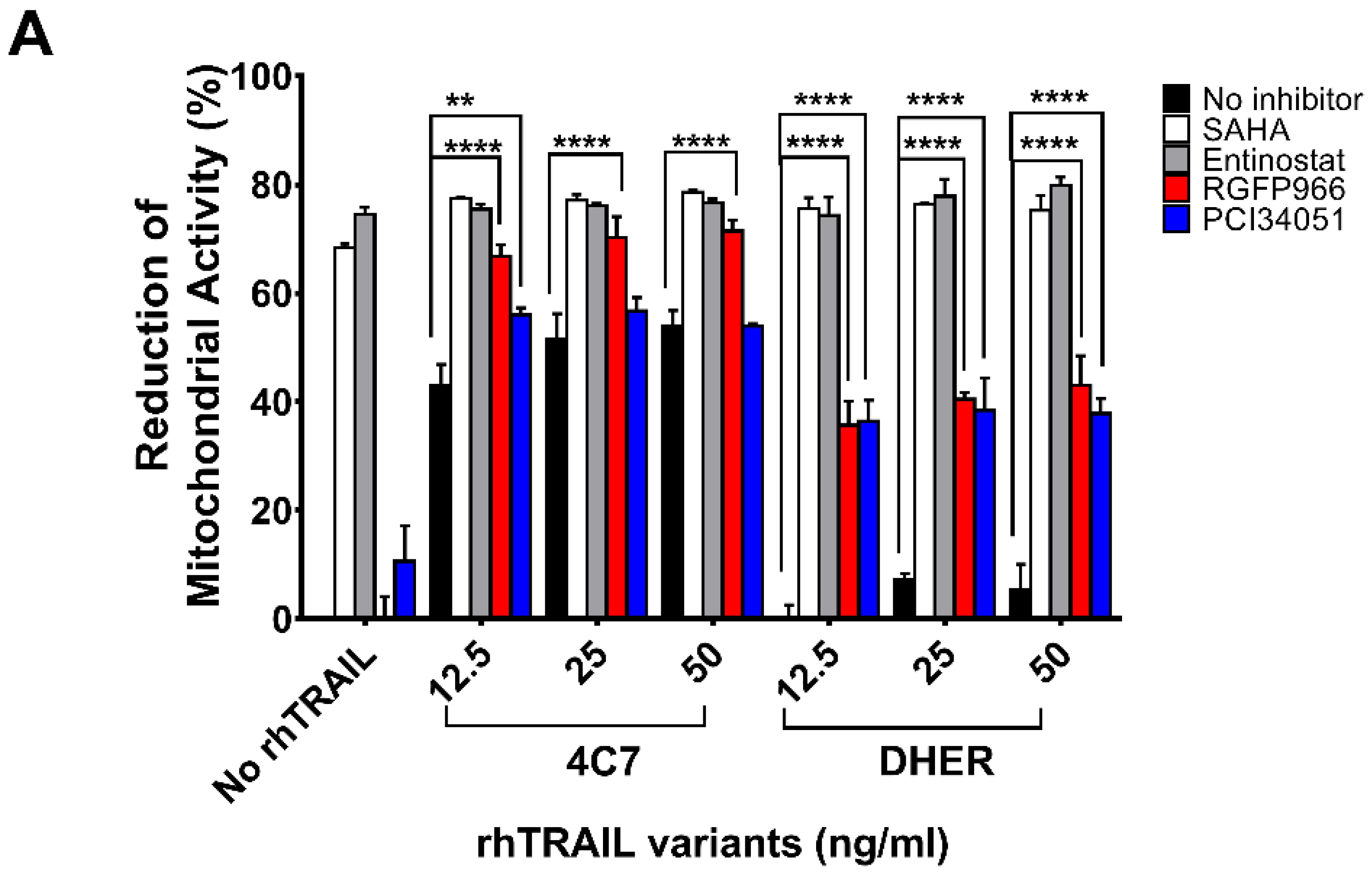
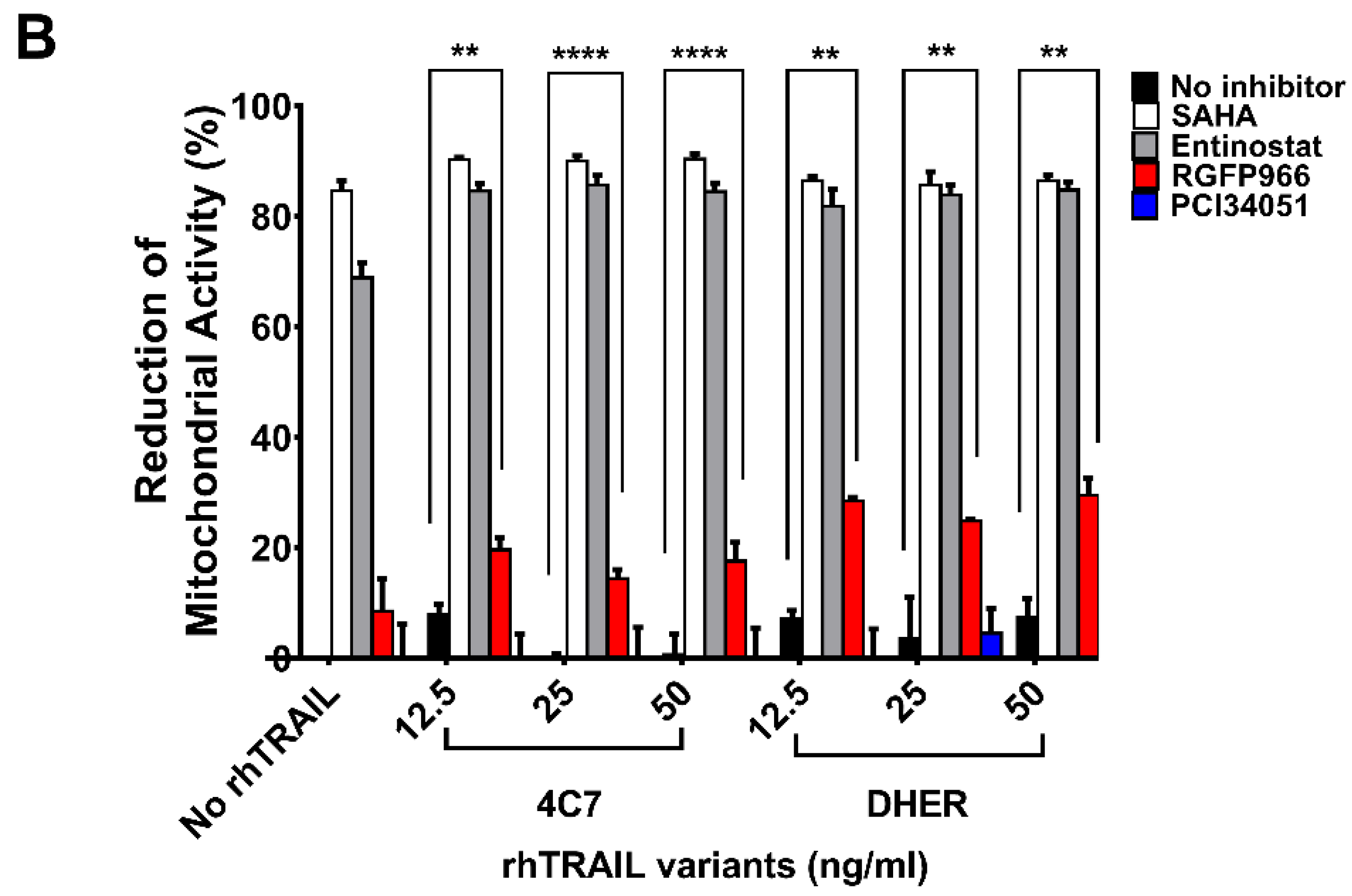
2.1. HDACi’s Enhance Cell Death in Combination with Receptor-Specific TRAIL Variants rhTRAIL 4C7 and DHER
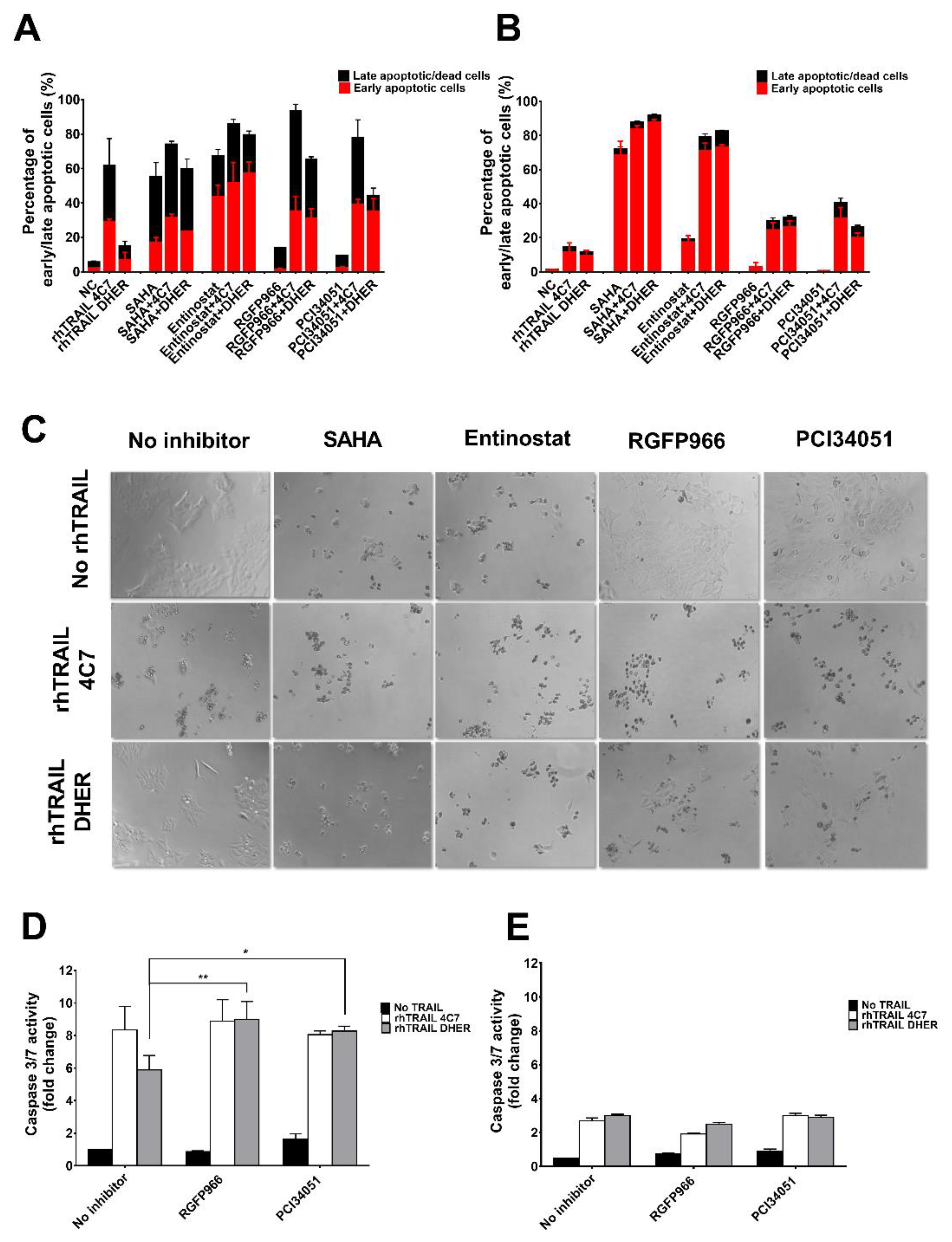
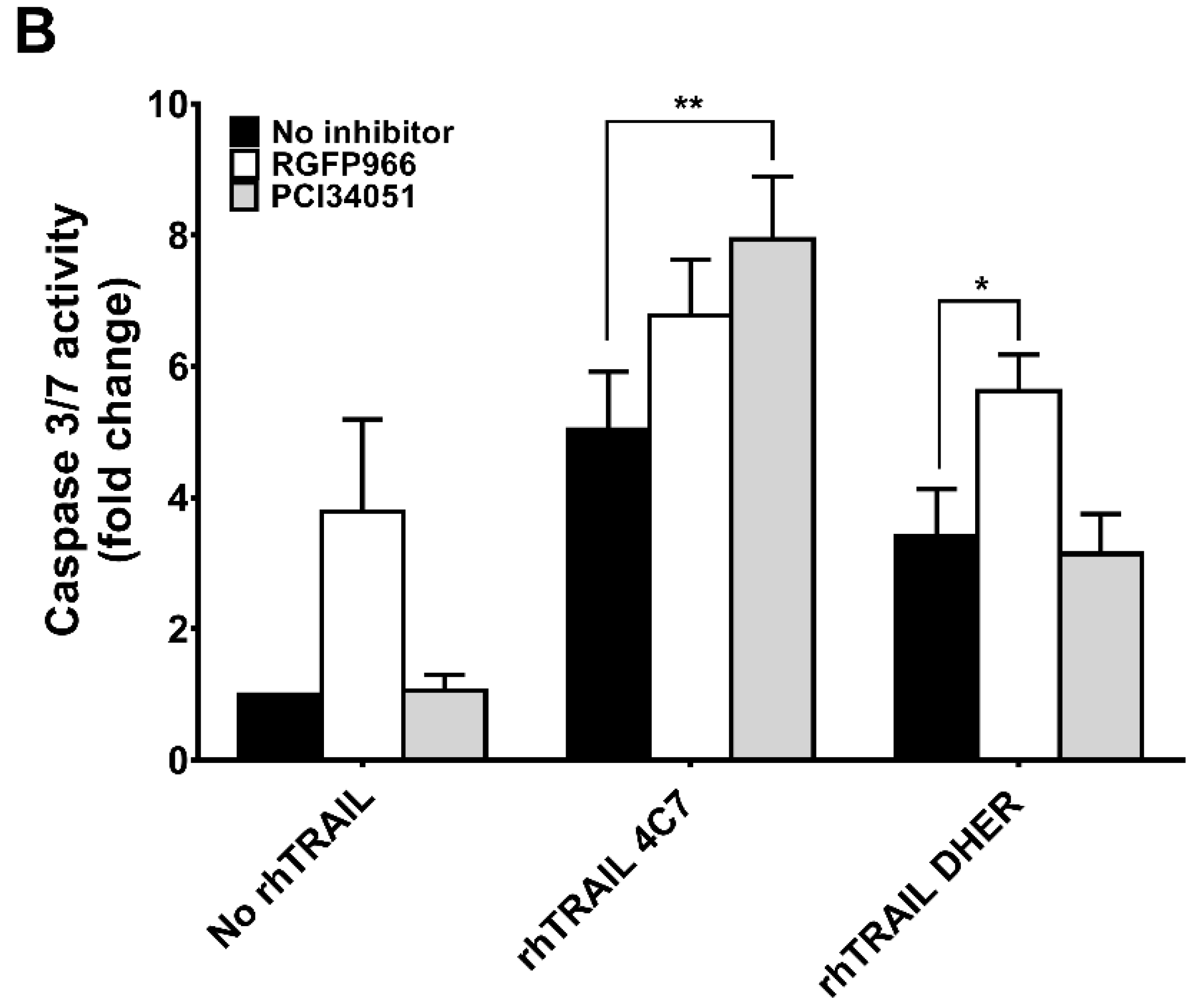
2.2. RGFP966 and PCI34051 Improve TRAIL-Induced Apoptosis
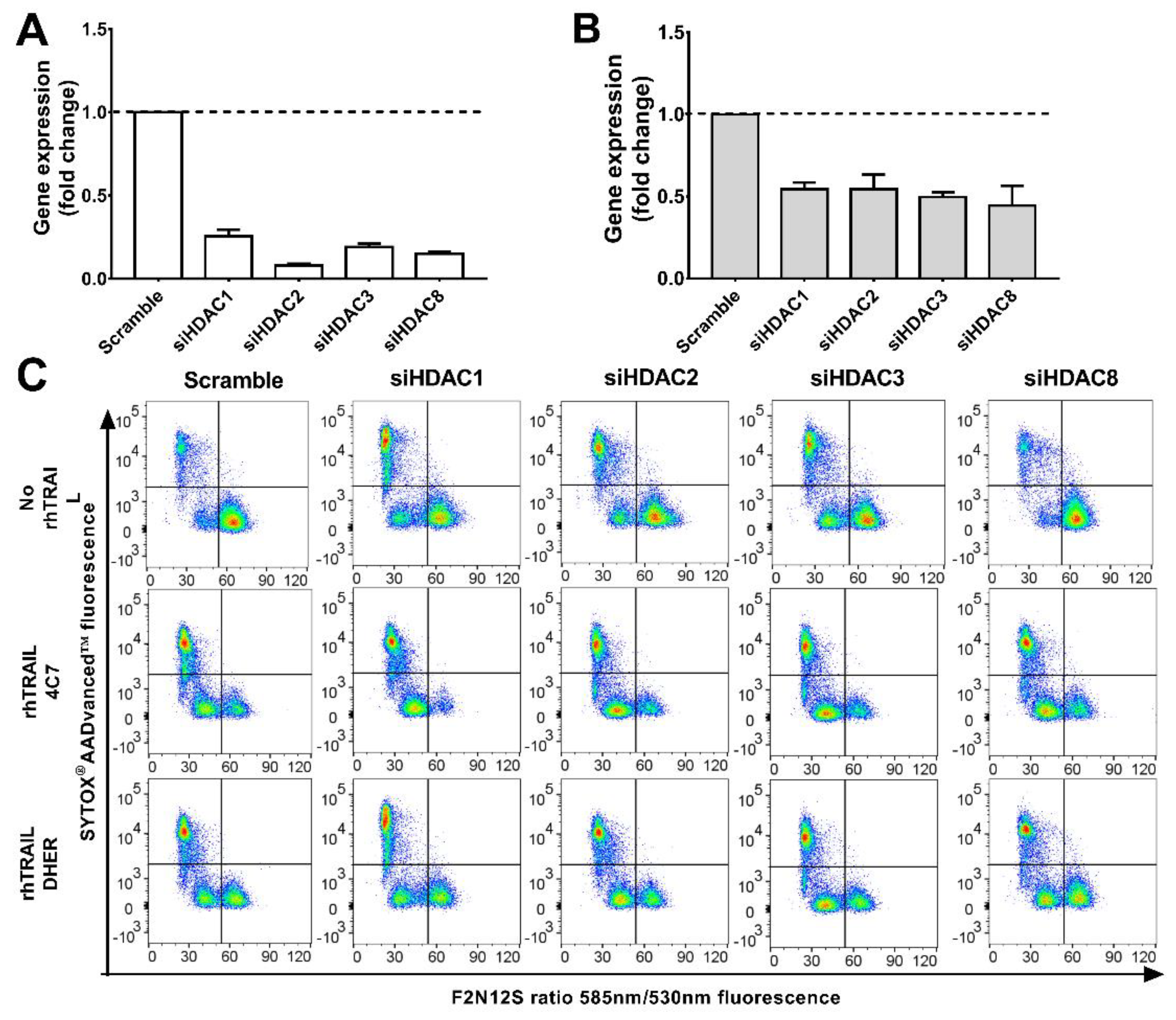
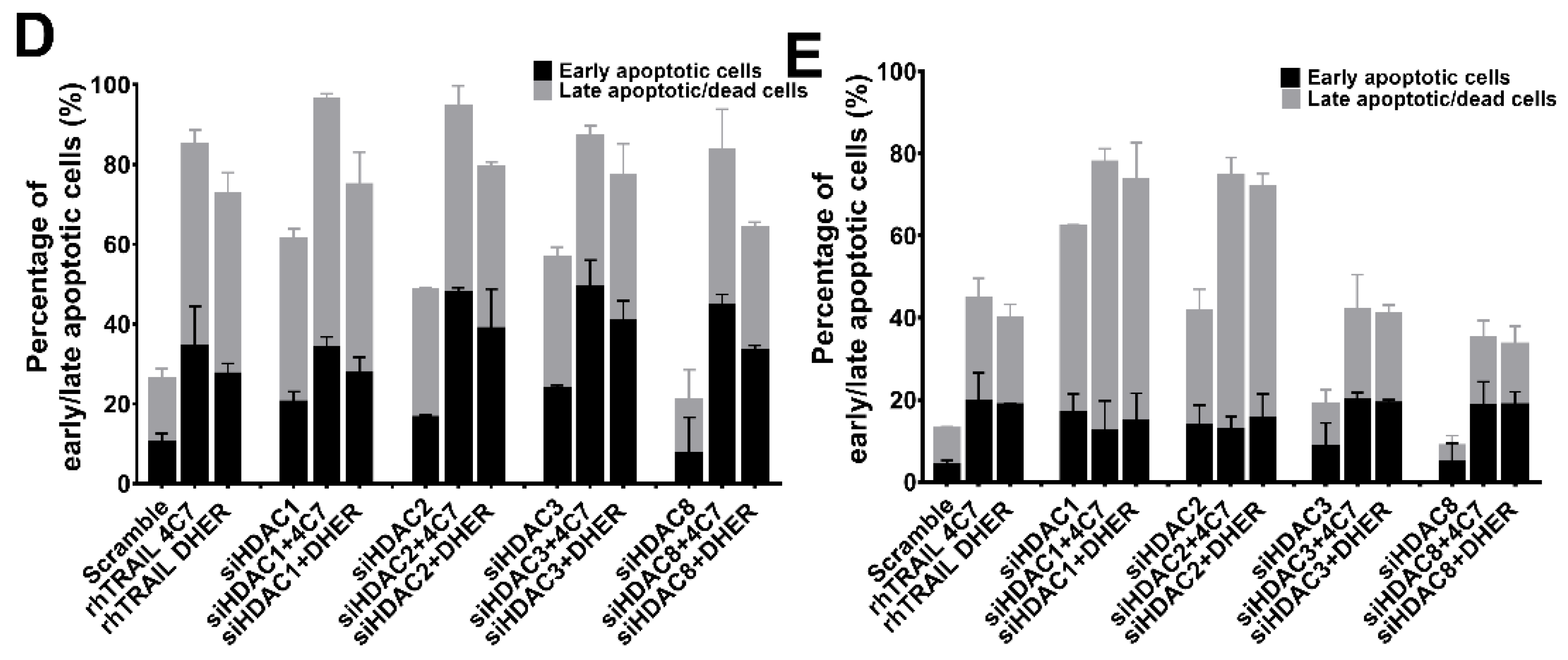
2.3. Knockdown of HDAC1, 2, 3, 8 Enhances TRAIL Sensitivity
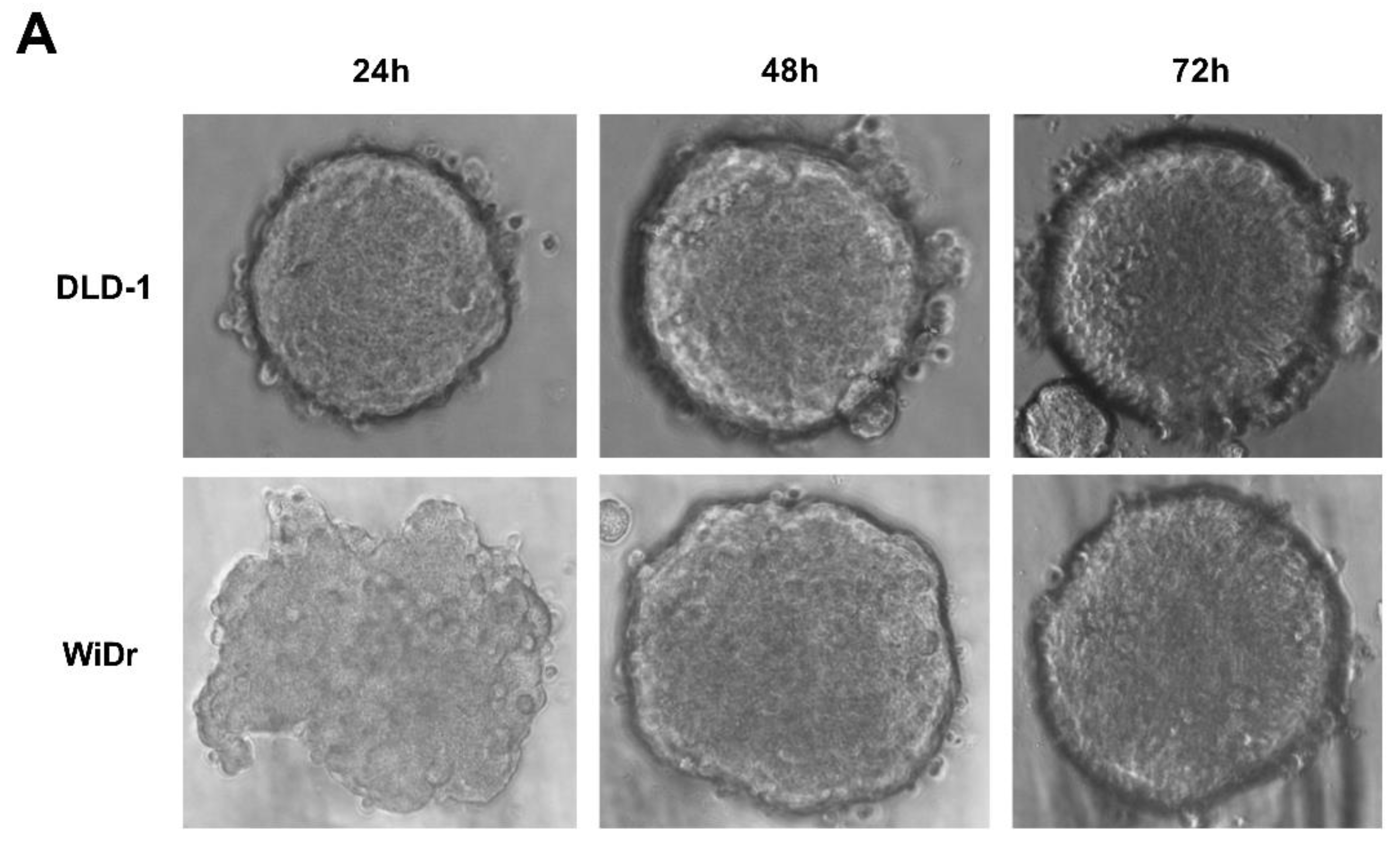
2.4. RGFP966 or PCI34051 Improves TRAIL-Induced Apoptosis in 3D Spheroid Model
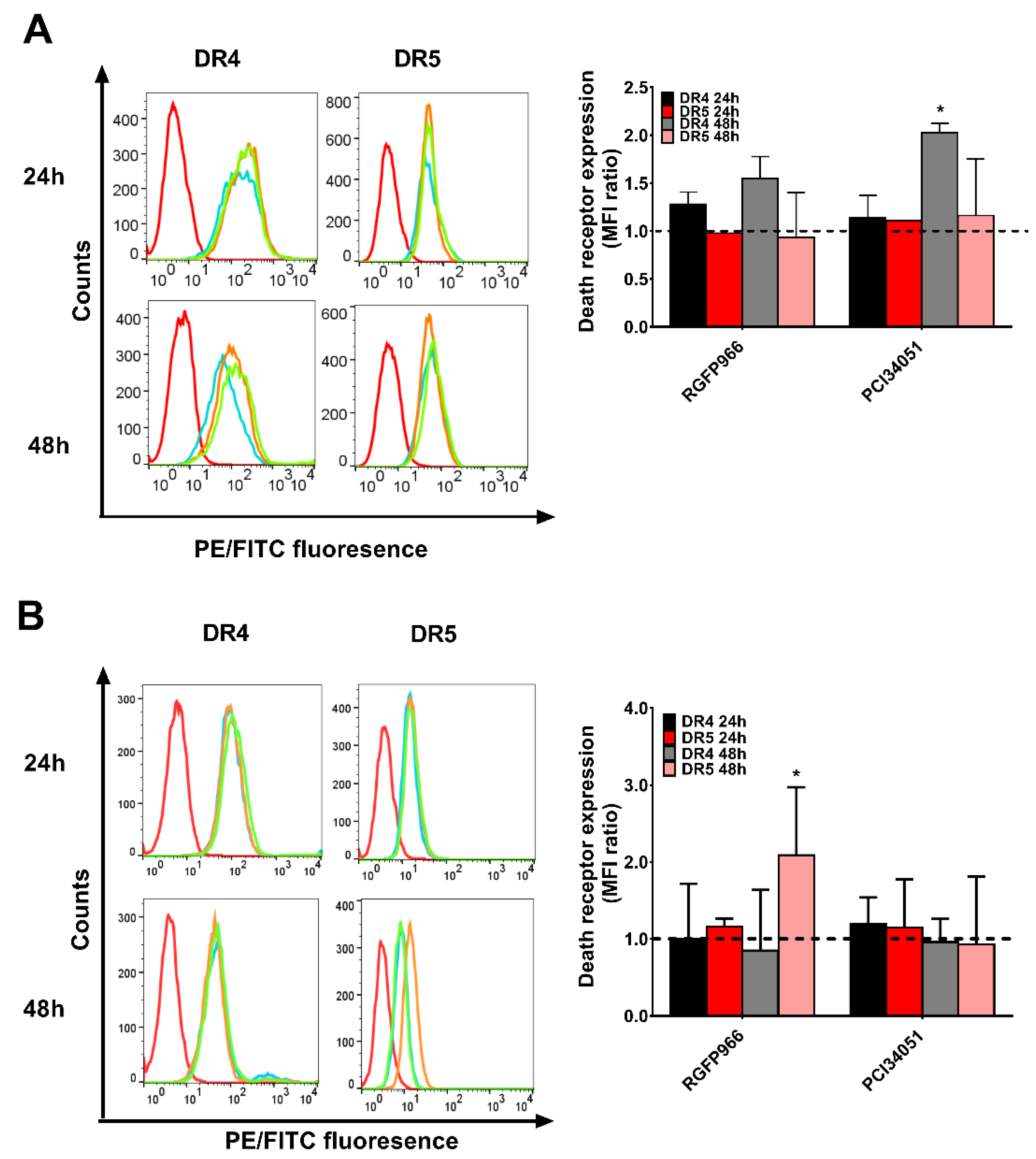
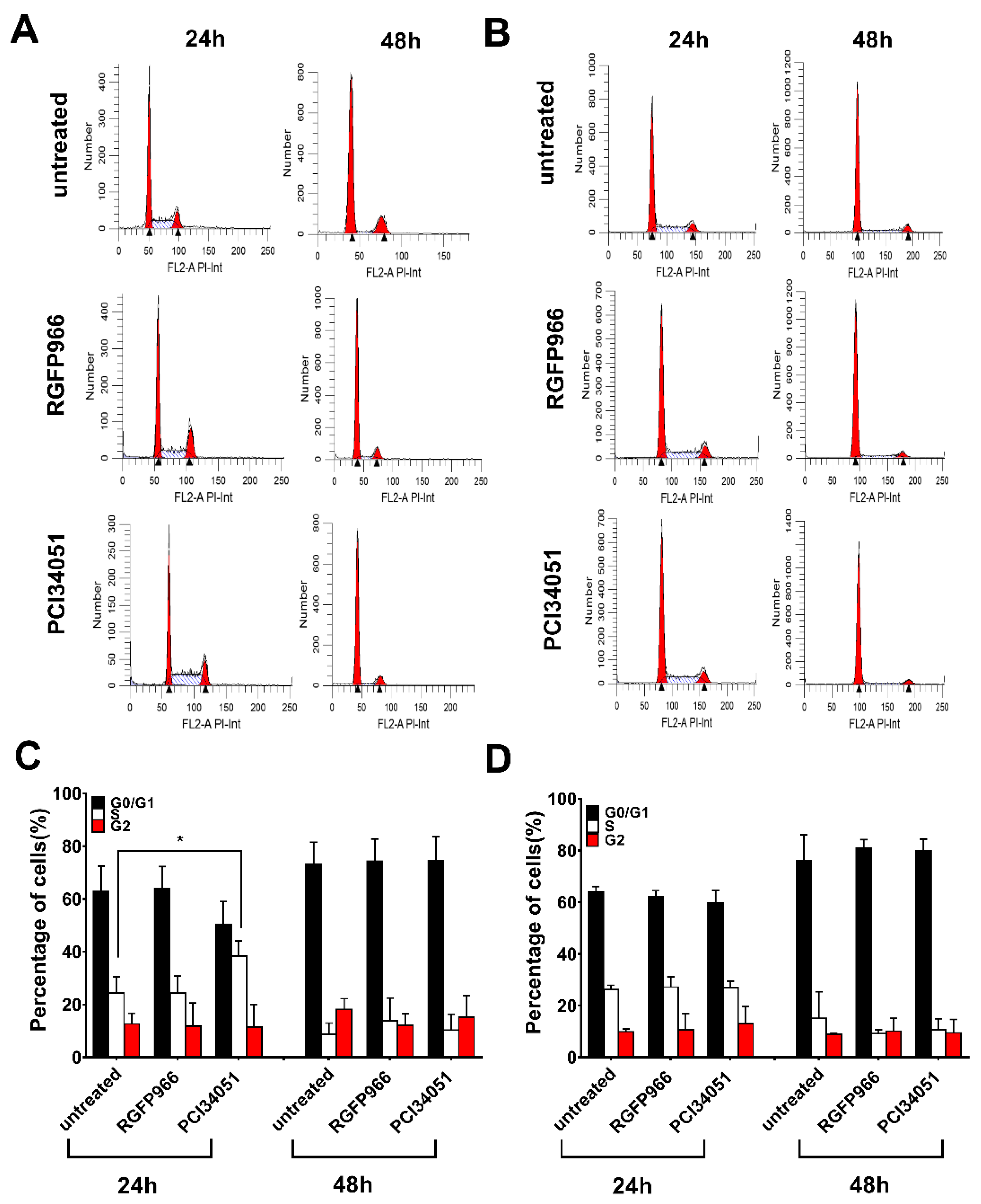
2.5. Expressions of Death Receptors and Cell Cycle Alter Upon HDACi’s Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cell Viability Assay
4.3. Apoptotic Assay
4.4. Caspase 3/7 Activity Assay
4.5. HDAC1, 2, 3, and 8 Knockdown Using siRNA
4.6. 3D Spheroid Construction
4.7. RNA Isolation and Quantitative Reverse Transcriptase PCR (qRT-PCR)
4.8. Death Receptor Expression Analysis
4.9. Cell Cycle Analysis
4.10. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; Mcmurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Pennarun, B.; Meijer, A.; de Vries, E.G.E.; Kleibeuker, J.H.; Kruyt, F.; de Jong, S. Playing the DISC: Turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim. Biophys. Acta 2010, 1805, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-κB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Kuang, A.A.; Diehl, G.E.; Zhang, J.; Winoto, A. FADD is required for DR4- and DR5-mediated apoptosis. Lack of trail-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J. Biol. Chem. 2000, 275, 25065–25068. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-Dependent Recruitment of Endogenous FADD and Caspase-8 to Death Receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Salvesen, G.S.; Ashkenazi, A. SnapShot: Caspases. Cell 2011, 147, 476. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Green, D.R. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostes, S.; Nettersheim, D.; Schorle, H. Epigenetic drugs and their molecular targets in testicular germ cell tumours. Nat. Rev. Urol. 2019, 16, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Spurling, C.C.; Godman, C.A.; Noonan, E.J.; Rasmussen, T.P.; Rosenberg, D.W.; Giardina, C. HDAC3 Overexpression and Colon Cancer Cell Proliferation and Differentiation. Mol. Carcinog. 2008, 47, 137–147. [Google Scholar] [CrossRef]
- Boix-Chornet, M.; Yamamoto, H.; Paz, M.F.; Aaltonen, L.A.; Schwartz, S.; Esteller, M.; Orntoft, T.F.; Setien, F.; Ballestar, E.; Alaminos, M.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar] [Green Version]
- Delhanty, J.D.A.; Wilson, A.; Kouzarides, T.; Ponder, B.A.J.; Linger, L.; Gayther, S.A.; Thorpe, K.; Daigo, Y.; Sowter, H.M.; Bannister, A.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar]
- Kishimoto, M.; Kohno, T.; Okudela, K.; Otsuka, A.; Sasaki, H.; Tanabe, C.; Sakiyama, T.; Hirama, C.; Kitabayashi, I.; Minna, J.D.; et al. Mutations and deletions of the CBP gene in human lung cancer. Clin. Cancer Res. 2005, 11, 512–519. [Google Scholar] [PubMed]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef]
- Shirakawa, K.; Chavez, L.; Hakre, S.; Calvanese, V.; Verdin, E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013, 21, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorff, S.; Li Shen, Y.; Mehrotra, N.; Pazdur, R.; Bullock, J.; Bloomquist, E.; Chen, X.-H.; Kwitkowski, V.E.; Kane, R.C.; Kaminskas, E.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [Green Version]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Pazdur, R.; Mann, B.S.; Johnson, J.R.; Justice, R.; Cohen, M.H. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [Green Version]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Heal. Drug Benefits 2016, 9, 84–87. [Google Scholar]
- De Graeff, P.; Laane, E.; Sjöberg, J.; Walsh, I.; Gisselbrecht, C.; Ludwig, H.; Pignatti, F.; Bergh, J.; Folin, A.; Tzogani, K.; et al. EMA Review of Panobinostat (Farydak) for the Treatment of Adult Patients with Relapsed and/or Refractory Multiple Myeloma. Oncologist 2017, 23, 631–636. [Google Scholar] [Green Version]
- Shah, J.J.; Feng, L.; Manasanch, E.E.; Weber, D.; Thomas, S.K.; Turturro, F.; Shah, N.; Popat, U.R.; Nieto, Y.; Bashir, Q.; et al. Phase I/II Trial of the Efficacy and Safety of Combination Therapy with Lenalidomide/Bortezomib/Dexamethasone (RVD) and Panobinostat in Transplant-Eligible Patients with Newly Diagnosed Multiple Myeloma. In Proceeding of 56th ASH Annual Meeing; American Association for Cancer Research: San Francisco, CA, USA, 2014; Volume 124, p. 33. [Google Scholar]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef]
- McLeod, A.B.; Stice, J.P.; Wardell, S.E.; Alley, H.M.; Chang, C.Y.; McDonnell, D.P. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2018, 78, 266–277. [Google Scholar] [CrossRef]
- Ramos, J.; Balasubramanian, S.; Buggy, J.J.; Verner, E.; Sirisawad, M.; Luo, W. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [Green Version]
- Aldana-masangkay, G.I.; Rodriguez-gonzalez, A.; Lin, T.; Ikeda, A.K.; Hsieh, Y.; Kim, Y.; Lomenick, B.; Okemoto, K.; Landaw, E.M.; Wang, D.P.; et al. Tubacin suppresses proliferation and induces apoptosis of acute lymphoblastic leukemia cells. Leuk Lymphoma 2014, 52, 1544–1555. [Google Scholar] [CrossRef]
- Reis, C.R.; van der Sloot, A.M.; Natoni, A.; Szegezdi, E.; Setroikromo, R.; Meijer, M.; Sjollema, K.; Stricher, F.; Cool, R.H.; Samali, A.; et al. Rapid and efficient cancer cell killing mediated by high-affinity death receptor homotrimerizing TRAIL variants. Cell Death Dis. 2010, 1, e83. [Google Scholar] [CrossRef]
- Van der Sloot, A.M.; Tur, V.; Szegezdi, E.; Mullally, M.M.; Cool, R.H.; Samali, A.; Serrano, L.; Quax, W.J. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8634–8639. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; Di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur. J. Cancer 2009, 45, 2425–2438. [Google Scholar] [CrossRef]
- Quast, S.A.; Steinhorst, K.; Plötz, M.; Eberle, J. Sensitization of Melanoma Cells for Death Ligand TRAIL Is Based on Cell Cycle Arrest, ROS Production, and Activation of Proapoptotic Bcl-2 Proteins. J. Investig. Dermatol. 2015, 135, 2794–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Saturno, G.; Valenti, M.; De Haven Brandon, A.; Thomas, G.V.; Eccles, S.; Clarke, P.A.; Workman, P. Combining trail with PI3 kinase or HSP90 inhibitors enhances apoptosis in colorectal cancer cells via suppression of survival signaling. Oncotarget 2013, 4, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Histone deacetylase (HDAC) inhibitors and regulation of TRAIL-induced apoptosis. Exp. Cell Res. 2012, 318, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Haecker, S.; Debatin, K.-M.; Fulda, S. Histone deacetylase inhibitors overcome resistance of caspase-8 negative cancers to TRAIL-induced apoptosis by upregulating caspase-8. In Proceedings of 98th AARG Annual Meeting; American Association for Cancer Research: Los Angeles, CA, USA, 2007; Volume 67, p. 702. [Google Scholar]
- Riley, J.S.; Hutchinson, R.; McArt, D.G.; Crawford, N.; Holohan, C.; Paul, I.; Van Schaeybroeck, S.; Salto-Tellez, M.; Johnston, P.G.; Fennell, D.A.; et al. Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer. Cell Death Dis. 2013, 4, e951. [Google Scholar] [CrossRef] [PubMed]
- Lauricella, M.; Ciraolo, A.; Carlisi, D.; Vento, R.; Tesoriere, G. SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231 and MCF-7 breast cancer cells. Biochimie 2012, 94, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Hektoen, M.; Ahmed, D.; Danielsen, S.A.; Lind, G.E.; Lothe, R.A.; Eide, P.W.; Eknæs, M.; Eilertsen, I.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef]
- Irvine, R.A.; Lin, I.G.; Hsieh, C.-L. DNA Methylation Has a Local Effect on Transcription and Histone Acetylation. Mol. Cell. Biol. 2002, 22, 6689–6696. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O. Regulation of TNF-related apoptosis-inducing ligand signaling by glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef] [PubMed]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morlé, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; van Roosmalen, I.A.M.; Reis, C.R.; Setroikromo, R.; Quax, W.J. Death receptor 5 is activated by fucosylation in colon cancer cells. FEBS J. 2019, 286, 555–571. [Google Scholar] [CrossRef]
- Montgomery, M.R.; Hull, E.E. Alterations in the glycome after HDAC inhibition impact oncogenic potential in epigenetically plastic SW13 cells. BMC Cancer 2019, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers 2019, 11, 645. https://doi.org/10.3390/cancers11050645
Zhang B, Liu B, Chen D, Setroikromo R, Haisma HJ, Quax WJ. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers. 2019; 11(5):645. https://doi.org/10.3390/cancers11050645
Chicago/Turabian StyleZhang, Baojie, Bin Liu, Deng Chen, Rita Setroikromo, Hidde J. Haisma, and Wim J. Quax. 2019. "Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells" Cancers 11, no. 5: 645. https://doi.org/10.3390/cancers11050645
APA StyleZhang, B., Liu, B., Chen, D., Setroikromo, R., Haisma, H. J., & Quax, W. J. (2019). Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers, 11(5), 645. https://doi.org/10.3390/cancers11050645