Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer

 and
and 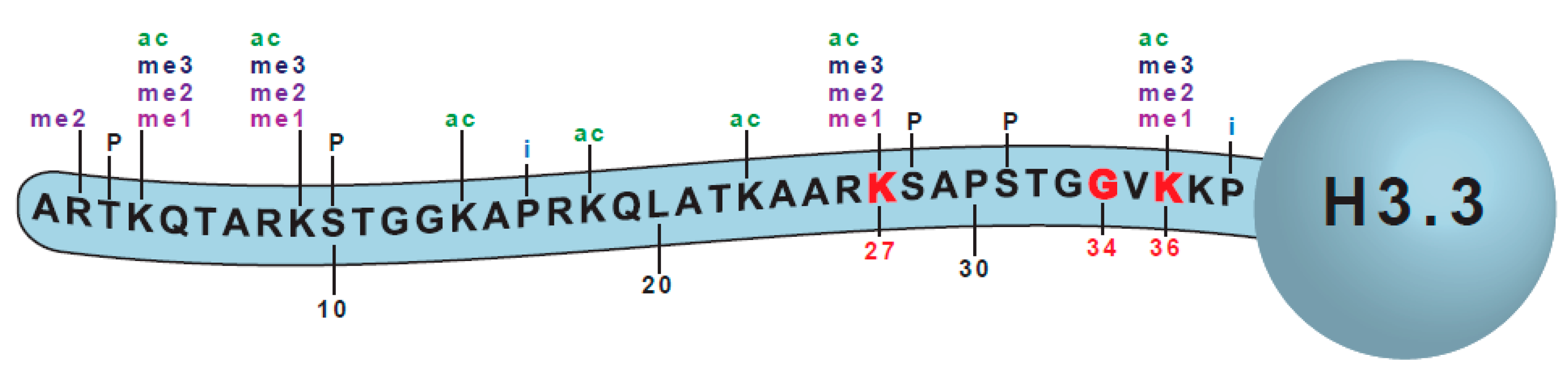{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
A Short Primer on Histone H3 Subtypes Relevant to This Review
2. The H3 K to M Mutants
2.1. K27M
2.1.1. Cancer Association of K27M
2.1.2. Effects on K27 Methylation
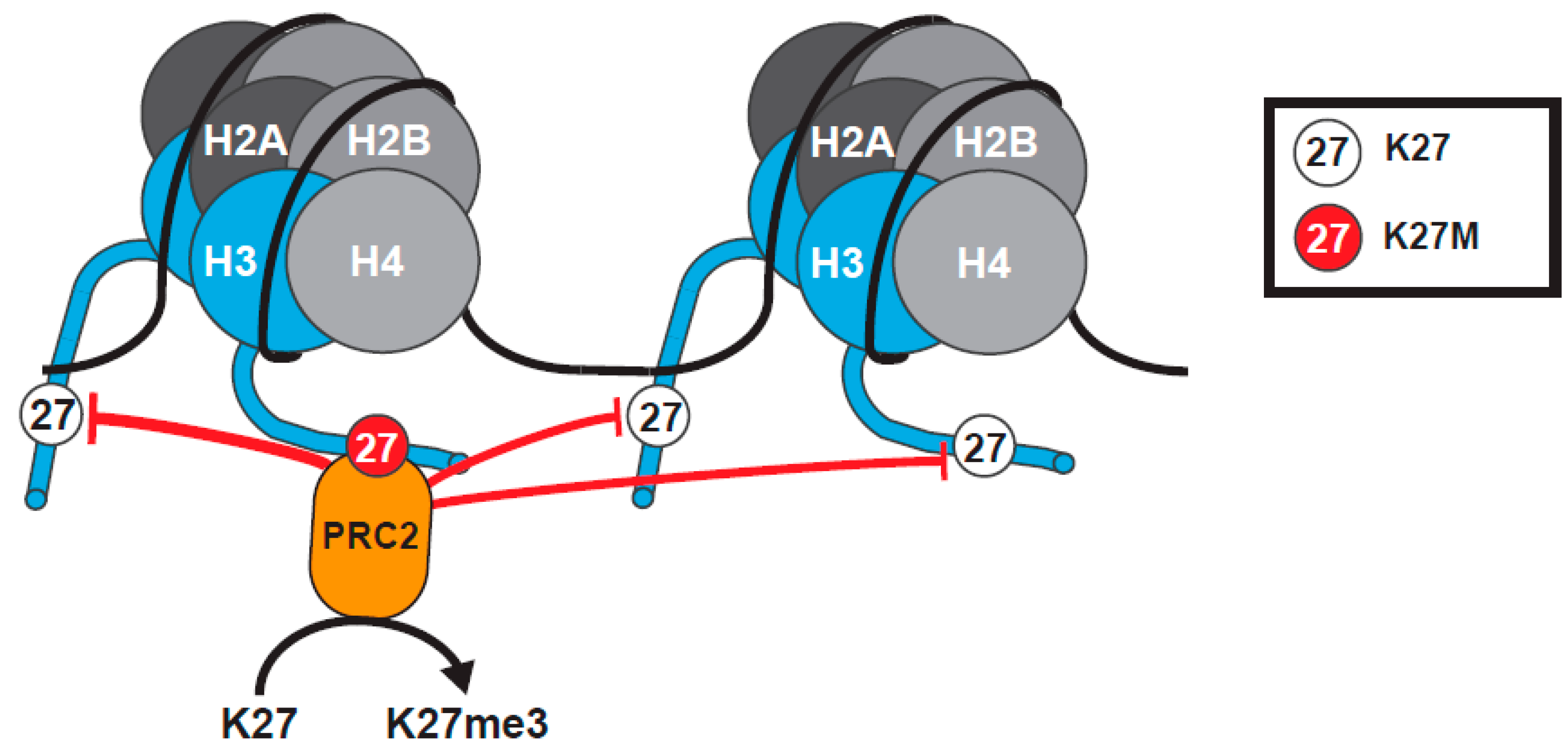
2.1.3. The Tethering and Sequestration Model
2.1.4. Recent Advances with Understanding H3K27M
2.2. K36M
2.2.1. Cancer Association of K36M
2.2.2. Effects on H3 K36 Methylation
2.2.3. Structural Considerations for K36M
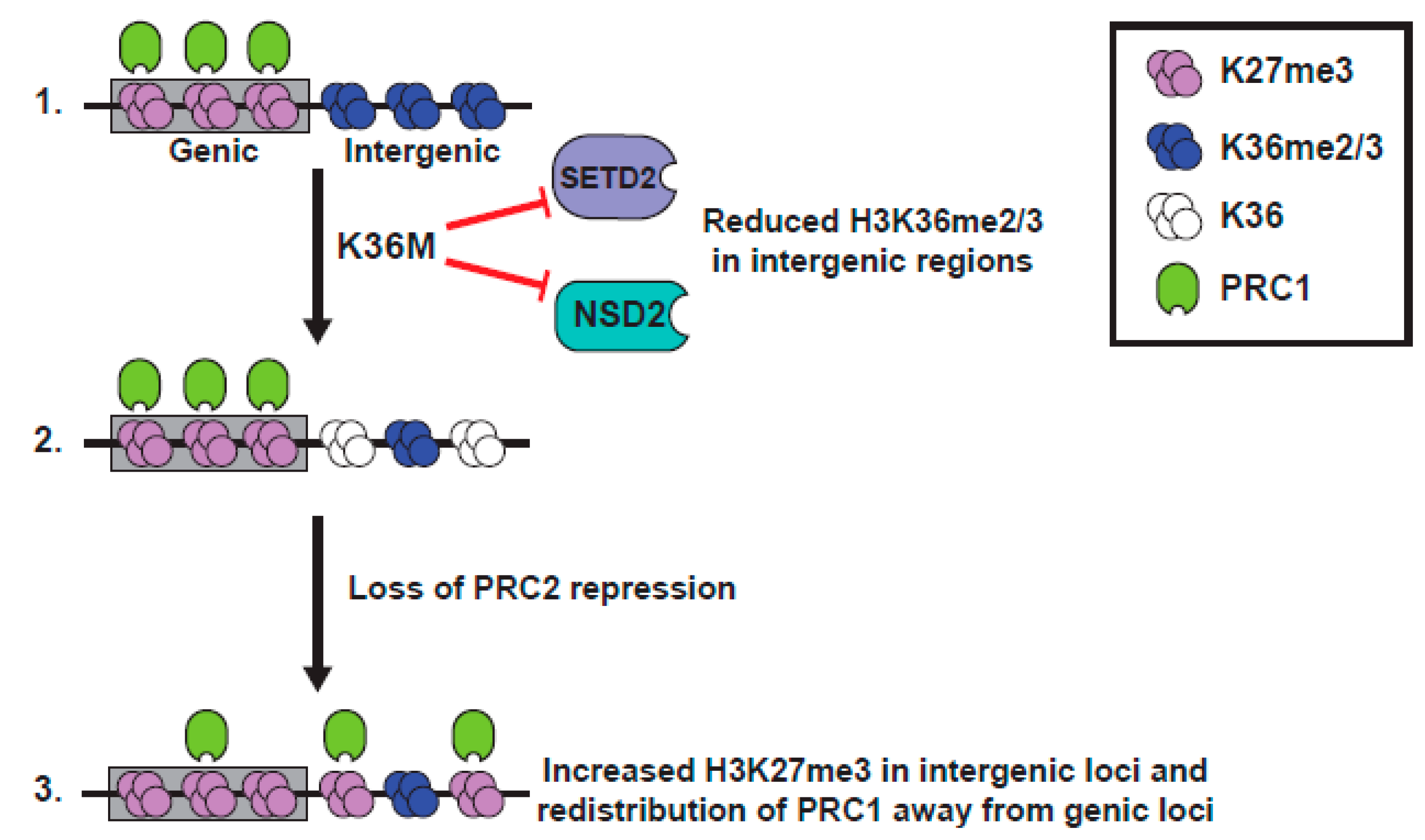
2.2.4. Biological Effects of K36M
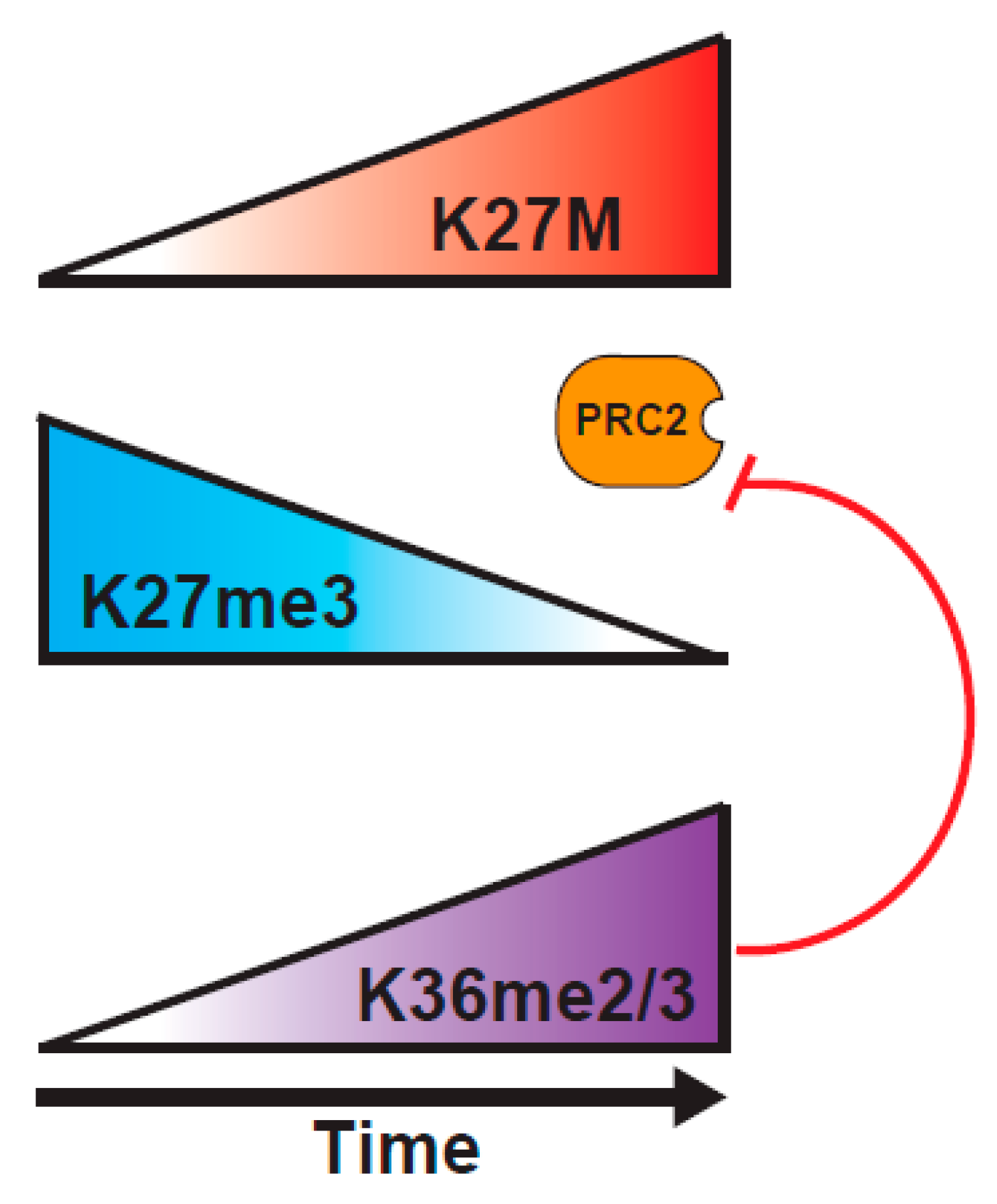
2.2.5. Interplay between K36M and K27 Methylation
3. The H3 G34 mutants
3.1. Cancer Association
3.2. Structural Considerations for G34 Mutants
3.3. Effects on H3K36 Methylation in G34 Mutant Backgrounds
3.4. Biological Effects of G34 Mutants
3.4.1. G34V
3.4.2. G34W/L
3.4.3. G34R
3.4.4. Does G34R Provide a New Source of Arg Methylation on the H3 Tail?
3.4.5. A Question of Dominance for G34R Mutants
4. Histone Genes Are Frequently Mutated in Adult as well as Pediatric Cancers
5. Conclusions and the Future for Histone Mutant Research in Cancer
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef]
- Shen, C.; Vakoc, C.R. Gain-of-function mutation of chromatin regulators as a tumorigenic mechanism and an opportunity for therapeutic intervention. Curr. Opin. Oncol. 2015, 27, 57–63. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [Green Version]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallee, V.P.; Hebert, J.; Sauvageau, G. H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef]
- Gessi, M.; Capper, D.; Sahm, F.; Huang, K.; von Deimling, A.; Tippelt, S.; Fleischhack, G.; Scherbaum, D.; Alfer, J.; Juhnke, B.O.; et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol. 2016, 132, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Donson, A.; Foreman, N.K.; Dorris, K. H3 K27M mutation in gangliogliomas can be associated with poor prognosis. Brain Pathol. 2017, 27, 846–850. [Google Scholar] [CrossRef]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Kallappagoudar, S.; Yadav, R.K.; Lowe, B.R.; Partridge, J.F. Histone H3 mutations-a special role for H3.3 in tumorigenesis? Chromosoma 2015, 124, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.S.; Tsai, S.; Bonner, W.M. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 1982, 31, 367–374. [Google Scholar] [CrossRef]
- Osley, M.A. The regulation of histone synthesis in the cell cycle. Annu. Rev. Biochem. 1991, 60, 827–861. [Google Scholar] [CrossRef]
- Schenk, R.; Jenke, A.; Zilbauer, M.; Wirth, S.; Postberg, J. H3.5 is a novel hominid-specific histone H3 variant that is specifically expressed in the seminiferous tubules of human testes. Chromosoma 2011, 120, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, S.M.; Mildner, S.N.; Bonisch, C.; Israel, L.; Maiser, A.; Matheisl, S.; Straub, T.; Merkl, R.; Leonhardt, H.; Kremmer, E.; et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell Biol. 2010, 190, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.K.; O’Day, K.; Trong, H.L.; Charbonneau, H.; Margolis, R.L. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc. Natl. Acad. Sci. USA 1991, 88, 3734–3738. [Google Scholar] [CrossRef]
- Franklin, S.G.; Zweidler, A. Non-allelic variants of histones 2a, 2b and 3 in mammals. Nature 1977, 266, 273–275. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.D.; George, A.J.; Morgan, K.A.; Mann, J.R.; Choo, K.H. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Filipescu, D.; Szenker, E.; Almouzni, G. Developmental roles of histone H3 variants and their chaperones. Trends Genet. 2013, 29, 630–640. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Ahmad, K. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005, 19, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.M.; Georgiou, A.; Szutorisz, H.; Maia e Silva, A.; Pombo, A.; Barahona, I.; Dargelos, E.; Canzonetta, C.; Dillon, N. Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 2005, 6, 354–360. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Zang, C.; Wei, G.; Cui, K.; Peng, W.; Zhao, K.; Felsenfeld, G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009, 41, 941–945. [Google Scholar] [CrossRef]
- Gaillard, P.H.; Martini, E.M.; Kaufman, P.D.; Stillman, B.; Moustacchi, E.; Almouzni, G. Chromatin assembly coupled to DNA repair: A new role for chromatin assembly factor I. Cell 1996, 86, 887–896. [Google Scholar] [CrossRef]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.; et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar] [CrossRef]
- Shibahara, K.; Stillman, B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999, 96, 575–585. [Google Scholar] [CrossRef]
- Gabrielli, F.; Aden, D.P.; Carrel, S.C.; von Bahr, C.; Rane, A.; Angeletti, C.A.; Hancock, R. Histone complements of human tissues, carcinomas, and carcinoma-derived cell lines. Mol. Cell Biochem. 1984, 65, 57–66. [Google Scholar] [CrossRef]
- Pina, B.; Suau, P. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev. Biol. 1987, 123, 51–58. [Google Scholar] [CrossRef]
- Frank, D.; Doenecke, D.; Albig, W. Differential expression of human replacement and cell cycle dependent H3 histone genes. Gene 2003, 312, 135–143. [Google Scholar] [CrossRef]
- Hake, S.B.; Garcia, B.A.; Duncan, E.M.; Kauer, M.; Dellaire, G.; Shabanowitz, J.; Bazett-Jones, D.P.; Allis, C.D.; Hunt, D.F. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 2006, 281, 559–568. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [Green Version]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pages, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broniscer, A.; Gajjar, A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: Two challenges for the pediatric oncologist. Oncologist 2004, 9, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated molecular meta-analysis of 1000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.S.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.; Choi, S.; Kim, D.S.; Choi, J.; Ha, Y.; Kim, K.N.; Suh, C.O.; Chang, J.H.; Kim, S.H.; Yoon, D.H. Impact of H3.3 K27M mutation on prognosis and survival of grade IV spinal cord glioma on the basis of new 2016 world health organization classification of the central nervous system. Neurosurgery 2019, 84, 1072–1081. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Tschiersch, B.; Hofmann, A.; Krauss, V.; Dorn, R.; Korge, G.; Reuter, G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994, 13, 3822–3831. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Garcia, B.A.; Shabanowitz, J.; Hunt, D.F. Characterization of histones and their post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2007, 11, 66–73. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; Van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Helin, K.; Dhanak, D. Chromatin proteins and modifications as drug targets. Nature 2013, 502, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Venneti, S.; Garimella, M.T.; Sullivan, L.M.; Martinez, D.; Huse, J.T.; Heguy, A.; Santi, M.; Thompson, C.B.; Judkins, A.R. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013, 23, 558–564. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 117. [Google Scholar] [CrossRef]
- Silveira, A.B.; Kasper, L.H.; Fan, Y.; Jin, H.; Wu, G.; Shaw, T.I.; Zhu, X.; Larson, J.D.; Easton, J.; Shao, Y.; et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019. [Google Scholar] [CrossRef]
- Jiao, L.; Liu, X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015, 350, aac4383. [Google Scholar] [CrossRef]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [Green Version]
- Brown, Z.Z.; Muller, M.M.; Jain, S.U.; Allis, C.D.; Lewis, P.W.; Muir, T.W. Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J. Am. Chem. Soc. 2014, 136, 13498–13501. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 2018, 35, 140–155. [Google Scholar] [CrossRef]
- Herz, H.M.; Morgan, M.; Gao, X.; Jackson, J.; Rickels, R.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014, 345, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Paucek, R.D.; Gooding, A.R.; Brown, Z.Z.; Ge, E.J.; Muir, T.W.; Cech, T.R. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol. 2017, 24, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Gan, H.; Cheng, L.; Lee, J.H.; Zhou, H.; Sarkaria, J.N.; Daniels, D.J.; Zhang, Z. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife 2018, 7, 36696. [Google Scholar] [CrossRef]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [Green Version]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3(K27M) cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 2017, 32, 684–700. [Google Scholar] [CrossRef]
- Stafford, J.M.; Lee, C.H.; Voigt, P.; Descostes, N.; Saldana-Meyer, R.; Yu, J.R.; LeRoy, G.; Oksuz, O.; Chapman, J.R.; Suarez, F.; et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 2018, 4, eaau5935. [Google Scholar] [CrossRef]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef]
- Tatavosian, R.; Duc, H.N.; Huynh, T.N.; Fang, D.; Schmitt, B.; Shi, X.; Deng, Y.; Phiel, C.; Yao, T.; Zhang, Z.; et al. Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stutzer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef]
- Wang, X.; Long, Y.; Paucek, R.D.; Gooding, A.R.; Lee, T.; Cech, T.R. Regulation of histone methylation by automethylation of PRC2. bioRxiv 2018, 343020. [Google Scholar] [CrossRef]
- Lee, C.H.; Granat, J.; Yu, J.R.; LeRoy, G.; Stafford, J.; Reinberg, D. Automethylation of PRC2 fine-tunes its catalytic activity on chromatin. bioRxiv 2018, 349449. [Google Scholar] [CrossRef]
- Baumhoer, D.; Amary, F.; Flanagan, A.M. An update of molecular pathology of bone tumors. Lessons learned from investigating samples by next generation sequencing. Genes Chromosomes Cancer 2019, 58, 88–99. [Google Scholar] [CrossRef]
- Yang, S.; Zheng, X.; Lu, C.; Li, G.M.; Allis, C.D.; Li, H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 2016, 30, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shan, C.M.; Wang, J.; Bao, K.; Tong, L.; Jia, S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci. Rep. 2017, 7, 43906. [Google Scholar] [CrossRef] [Green Version]
- Jayaram, H.; Hoelper, D.; Jain, S.U.; Cantone, N.; Lundgren, S.M.; Poy, F.; Allis, C.D.; Cummings, R.; Bellon, S.; Lewis, P.W. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc. Natl. Acad. Sci. USA 2016, 113, 6182–6187. [Google Scholar] [CrossRef]
- Reznikoff, C.A.; Bertram, J.S.; Brankow, D.W.; Heidelberger, C. Quantitative and qualitative studies of chemical transformation of cloned C3H mouse embryo cells sensitive to postconfluence inhibition of cell division. Cancer Res. 1973, 33, 3239–3249. [Google Scholar]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Werner, M. Giant cell tumour of bone: Morphological, biological and histogenetical aspects. Int. Orthop. 2006, 30, 484–489. [Google Scholar] [CrossRef]
- Toledo, R.A.; Qin, Y.; Cheng, Z.M.; Gao, Q.; Iwata, S.; Silva, G.M.; Prasad, M.L.; Ocal, I.T.; Rao, S.; Aronin, N.; et al. Recurrent mutations of chromatin-remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin. Cancer Res. 2016, 22, 2301–2310. [Google Scholar] [CrossRef]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef]
- Cheng, Z.; Cheung, P.; Kuo, A.J.; Yukl, E.T.; Wilmot, C.M.; Gozani, O.; Patel, D.J. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev. 2014, 28, 1758–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.J.; Santos-Rosa, H.; Kouzarides, T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 2006, 126, 905–916. [Google Scholar] [CrossRef]
- Shi, L.; Shi, J.; Shi, X.; Li, W.; Wen, H. Histone H3.3 G34 mutations alter histone H3K36 and H3K27 methylation in cis. J. Mol. Biol. 2018, 430, 1562–1565. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Udugama, M.; Lin, W.; Hii, L.; Law, R.H.P.; Steer, D.L.; Das, P.P.; Mann, J.R.; Wong, L.H. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun. 2018, 9, 3142. [Google Scholar] [CrossRef]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef]
- Lim, J.; Park, J.H.; Baude, A.; Yoo, Y.; Lee, Y.K.; Schmidt, C.R.; Park, J.B.; Fellenberg, J.; Zustin, J.; Haller, F.; et al. The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci. Rep. 2017, 7, 13459. [Google Scholar] [CrossRef] [Green Version]
- Sidoli, S.; Garcia, B.A. Middle-down proteomics: A still unexploited resource for chromatin biology. Expert Rev. Proteom. 2017, 14, 617–626. [Google Scholar] [CrossRef]
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.A.; Shibata, Y.; Noma, K.; Tsukamoto, Y.; Warren, E.; Temple, B.; Grewal, S.I.; Strahl, B.D. Histone H3 K36 methylation is associated with transcription elongation in Schizosaccharomyces pombe. Eukaryot. Cell 2005, 4, 1446–1454. [Google Scholar] [CrossRef]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef]
- Molina-Serrano, D.; Schiza, V.; Kirmizis, A. Cross-talk among epigenetic modifications: Lessons from histone arginine methylation. Biochem. Soc. Trans. 2013, 41, 751–759. [Google Scholar] [CrossRef]
- Guccione, E.; Bassi, C.; Casadio, F.; Martinato, F.; Cesaroni, M.; Schuchlautz, H.; Luscher, B.; Amati, B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007, 449, 933–937. [Google Scholar] [CrossRef]
- Hyllus, D.; Stein, C.; Schnabel, K.; Schiltz, E.; Imhof, A.; Dou, Y.; Hsieh, J.; Bauer, U.M. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007, 21, 3369–3380. [Google Scholar] [CrossRef] [Green Version]
- Kirmizis, A.; Santos-Rosa, H.; Penkett, C.J.; Singer, M.A.; Vermeulen, M.; Mann, M.; Bahler, J.; Green, R.D.; Kouzarides, T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 2007, 449, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Yagoub, D.; Hart-Smith, G.; Moecking, J.; Erce, M.A.; Wilkins, M.R. Yeast proteins Gar1p, Nop1p, Npl3p, Nsr1p, and Rps2p are natively methylated and are substrates of the arginine methyltransferase Hmt1p. Proteomics 2015, 15, 3209–3218. [Google Scholar] [CrossRef]
- Low, J.K.; Im, H.; Erce, M.A.; Hart-Smith, G.; Snyder, M.P.; Wilkins, M.R. Protein substrates of the arginine methyltransferase Hmt1 identified by proteome arrays. Proteomics 2016, 16, 465–476. [Google Scholar] [CrossRef]
- Bachand, F.; Silver, P.A. PRMT3 is a ribosomal protein methyltransferase that affects the cellular levels of ribosomal subunits. EMBO J. 2004, 23, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Gary, J.D.; Lin, W.J.; Yang, M.C.; Herschman, H.R.; Clarke, S. The predominant protein-arginine methyltransferase from Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 12585–12594. [Google Scholar] [CrossRef]
- Pawlak, M.R.; Scherer, C.A.; Chen, J.; Roshon, M.J.; Ruley, H.E. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell Biol. 2000, 20, 4859–4869. [Google Scholar] [CrossRef]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar] [CrossRef]
- Chen, Z.; Zang, J.; Whetstine, J.; Hong, X.; Davrazou, F.; Kutateladze, T.G.; Simpson, M.; Mao, Q.; Pan, C.H.; Dai, S.; et al. Structural insights into histone demethylation by JMJD2 family members. Cell 2006, 125, 691–702. [Google Scholar] [CrossRef]
- Couture, J.F.; Collazo, E.; Ortiz-Tello, P.A.; Brunzelle, J.S.; Trievel, R.C. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat. Struct. Mol. Biol. 2007, 14, 689–695. [Google Scholar] [CrossRef]
- Meyronet, D.; Esteban-Mader, M.; Bonnet, C.; Joly, M.O.; Uro-Coste, E.; Amiel-Benouaich, A.; Forest, F.; Rousselot-Denis, C.; Burel-Vandenbos, F.; Bourg, V.; et al. Characteristics of H3 K27M-mutant gliomas in adults. Neuro-Oncology 2017, 19, 1127–1134. [Google Scholar] [CrossRef]
- Aihara, K.; Mukasa, A.; Gotoh, K.; Saito, K.; Nagae, G.; Tsuji, S.; Tatsuno, K.; Yamamoto, S.; Takayanagi, S.; Narita, Y.; et al. H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro-Oncology 2014, 16, 140–146. [Google Scholar] [CrossRef]
- Feng, J.; Hao, S.; Pan, C.; Wang, Y.; Wu, Z.; Zhang, J.; Yan, H.; Zhang, L.; Wan, H. The H3.3 K27M mutation results in a poorer prognosis in brainstem gliomas than thalamic gliomas in adults. Hum. Pathol. 2015, 46, 1626–1632. [Google Scholar] [CrossRef]
- Gessi, M.; Gielen, G.H.; Dreschmann, V.; Waha, A.; Pietsch, T. High frequency of H3F3A (K27M) mutations characterizes pediatric and adult high-grade gliomas of the spinal cord. Acta Neuropathol. 2015, 130, 435–437. [Google Scholar] [CrossRef]
- Reyes-Botero, G.; Giry, M.; Mokhtari, K.; Labussiere, M.; Idbaih, A.; Delattre, J.Y.; Laigle-Donadey, F.; Sanson, M. Molecular analysis of diffuse intrinsic brainstem gliomas in adults. J. Neurooncol. 2014, 116, 405–411. [Google Scholar] [CrossRef]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.; Perry, A. Diffuse midline gliomas with histone H3-K27M mutation: A series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef]
- Collord, G.; Martincorena, I.; Young, M.D.; Foroni, L.; Bolli, N.; Stratton, M.R.; Vassiliou, G.S.; Campbell, P.J.; Behjati, S. Recurrent histone mutations in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2019, 184, 676–679. [Google Scholar] [CrossRef]
- Atak, Z.K.; Gianfelici, V.; Hulselmans, G.; De Keersmaecker, K.; Devasia, A.G.; Geerdens, E.; Mentens, N.; Chiaretti, S.; Durinck, K.; Uyttebroeck, A.; et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013, 9, e1003997. [Google Scholar] [CrossRef]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.A.; Liu, X.Y.; Sturm, D.; Korshunov, A.; Jones, D.T.; Witt, H.; Kool, M.; Albrecht, S.; et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.K.; Ai, H.S.; Li, Y.M.; Yan, B. Total chemical synthesis of modified histones. Front Chem. 2018, 6, 19. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. https://doi.org/10.3390/cancers11050660
Lowe BR, Maxham LA, Hamey JJ, Wilkins MR, Partridge JF. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers. 2019; 11(5):660. https://doi.org/10.3390/cancers11050660
Chicago/Turabian StyleLowe, Brandon R., Lily A. Maxham, Joshua J. Hamey, Marc R. Wilkins, and Janet F. Partridge. 2019. "Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer" Cancers 11, no. 5: 660. https://doi.org/10.3390/cancers11050660
APA StyleLowe, B. R., Maxham, L. A., Hamey, J. J., Wilkins, M. R., & Partridge, J. F. (2019). Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers, 11(5), 660. https://doi.org/10.3390/cancers11050660