Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer



Abstract
:1. Introduction
2. β1 Integrins
2.1. β1 Integrins Promote Resistance to EGFR-Targeted Therapies
2.1.1. Cooperation between β1 Integrin and EGFR in Cancer Cells
2.1.2. Molecular Mechanism of β1 Integrin-Mediated Resistance to EGFR-Targeted Therapies
2.2. B1 Integrins Promote Resistance to Therapies Targeting HER2
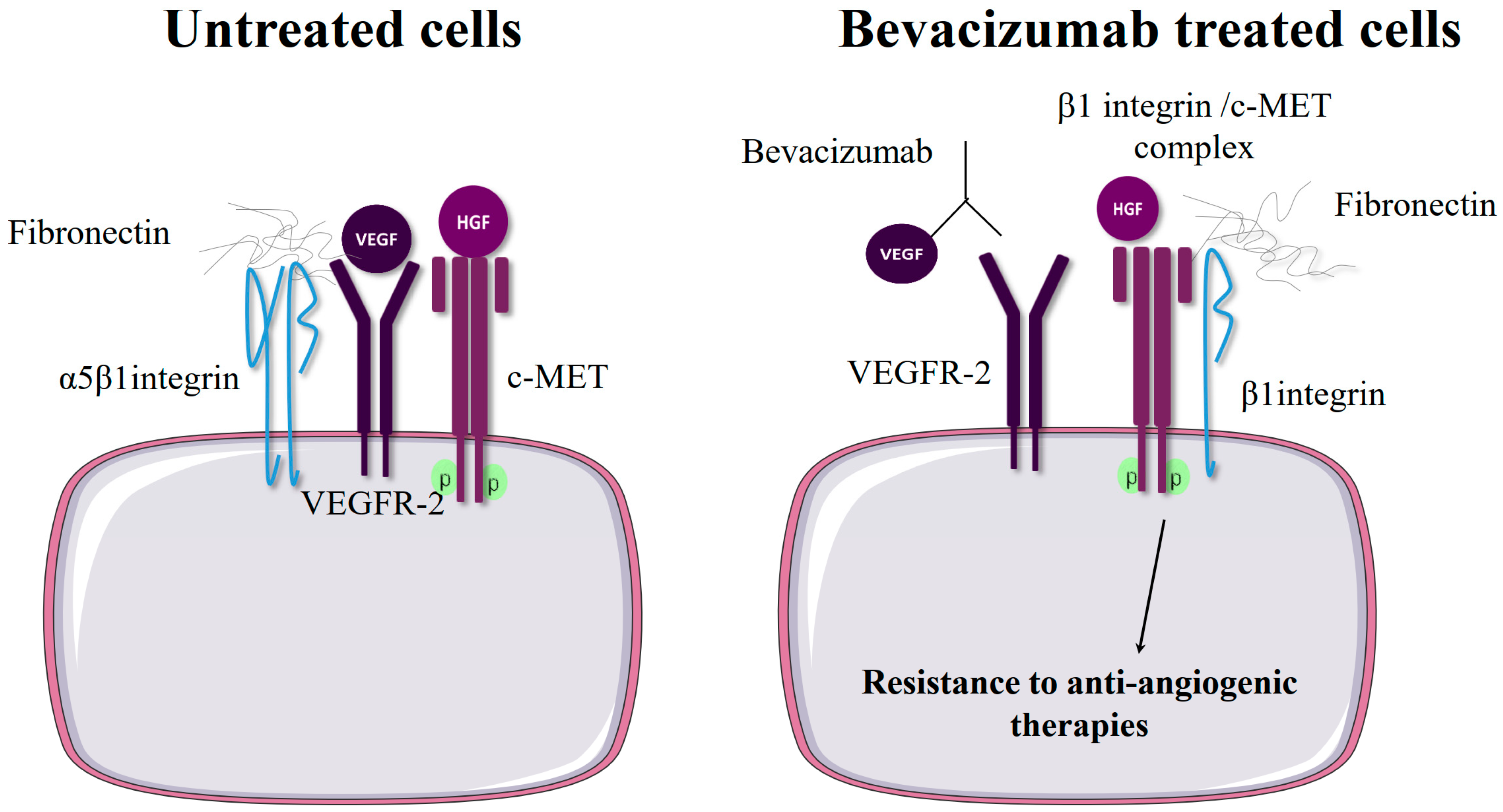
2.3. β1 Integrin Expression Confers Resistance to Anti-Angiogenic Therapies Targeting VEGFR or c-Met
3. αvβ Integrins
3.1. αv Integrin Triggers Resistance to Anti-EGFR Therapies
3.2. αvβ3 Integrin Involvement in Resistance to Drugs Targeting Other RTKs
4. α6β4 Integrins
4.1. Crosstalk between α6β4 Integrin and Growth Factor Receptors
4.2. α6β4 Integrin and Resistance to Anti-HER2 Therapies
5. Integrins and Carcinoma-Associated Fibroblasts
6. Conclusions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.T.; Smalley, K.S.M. Fibroblast-mediated drug resistance in cancer. Biochem. Pharmacol. 2013, 85, 1033–1041. [Google Scholar] [CrossRef]
- Zoeller, J.J.; Bronson, R.T.; Selfors, L.M.; Mills, G.B.; Brugge, J.S. Niche-localized tumor cells are protected from HER2-targeted therapy via upregulation of an anti-apoptotic program in vivo. NPJ Breast Cancer 2017, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 2011, 27, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef]
- Xing, Z.; Chen, H.C.; Nowlen, J.K.; Taylor, S.J.; Shalloway, D.; Guan, J.L. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol. Biol. Cell 1994, 5, 413–421. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Park, M. Receptor tyrosine kinases: Role in cancer progression. Curr. Oncol. 2006, 13, 191–193. [Google Scholar]
- Eke, I.; Storch, K.; Krause, M.; Cordes, N. Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer Res. 2013, 73, 5869–5879. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Jung, K.; Baek, D.-S.; Hong, S.-S.; Kim, Y.-S. Co-targeting of EGF receptor and neuropilin-1 overcomes cetuximab resistance in pancreatic ductal adenocarcinoma with integrin β1-driven Src-Akt bypass signaling. Oncogene 2017, 36, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, S.K.; Li, X. Integrin α5/β1 Mediates fibronectin-dependent epithelial cell proliferation through epidermal growth factor receptor activation. Mol. Biol. Cell 2000, 11, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
- Kanda, R.; Kawahara, A.; Watari, K.; Murakami, Y.; Sonoda, K.; Maeda, M.; Fujita, H.; Kage, M.; Uramoto, H.; Costa, C.; et al. Erlotinib resistance in lung cancer cells mediated by integrin β1/Src/Akt-driven bypass signaling. Cancer Res. 2013, 73, 6243–6253. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, M.; Das, S.; Berns, E.J.; Kim, J.; Stupp, S.I.; Kessler, J.A. Nanofiber-mediated inhibition of focal adhesion kinase sensitizes glioma stemlike cells to epidermal growth factor receptor inhibition. Neuro. Oncol. 2013, 15, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.-C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011, 13, R84. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Estrada, M.V.; Bianchini, G.; Moore, P.D.; Zhao, J.; Cheng, F.; Koch, J.P.; Gianni, L.; Tyson, D.R.; Sánchez, V.; et al. Extracellular matrix/integrin signaling promotes resistance to combined inhibition of HER2 and PI3K in HER2+ breast cancer. Cancer Res 2017, 77, 3280–3292. [Google Scholar] [CrossRef]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; Nguyen, A.; Chandra, A.; Sidorov, M.K.; Yagnik, G.; Rick, J.; Han, S.W.; Chen, W.; Flanigan, P.M.; Schneidman-Duhovny, D.; et al. Cross-activating c-Met/β1 integrin complex drives metastasis and invasive resistance in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E8685–E8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef]
- Yue, J.; Lv, D.; Wang, C.; Li, L.; Zhao, Q.; Chen, H.; Xu, L. Epigenetic silencing of miR-483-3p promotes acquired gefitinib resistance and EMT in EGFR-mutant NSCLC by targeting integrin β3. Oncogene 2018, 37, 4300–4312. [Google Scholar] [CrossRef]
- Shin, D.H.; Lee, H.-J.; Min, H.-Y.; Choi, S.P.; Lee, M.-S.; Lee, J.W.; Johnson, F.M.; Mehta, K.; Lippman, S.M.; Glisson, B.S.; et al. Combating resistance to anti-IGFR antibody by targeting the integrin β3-Src pathway. J. Natl. Cancer Inst. 2013, 105, 1558–1570. [Google Scholar] [CrossRef]
- Yi, H.; Zeng, D.; Shen, Z.; Liao, J.; Wang, X.; Liu, Y.; Zhang, X.; Kong, P. Integrin alphavbeta3 enhances β-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: Implications for microenvironment influence on sorafenib sensitivity. Oncotarget 2016, 7, 40387–40397. [Google Scholar] [CrossRef]
- Zhang, P.-F.; Li, K.-S.; Shen, Y.; Gao, P.-T.; Dong, Z.-R.; Cai, J.-B.; Zhang, C.; Huang, X.-Y.; Tian, M.-X.; Hu, Z.-Q.; et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016, 7, e2201. [Google Scholar] [CrossRef]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. β4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef]
- Yang, X.H.; Flores, L.M.; Li, Q.; Zhou, P.; Xu, F.; Krop, I.E.; Hemler, M.E. Disruption of laminin-integrin-CD151-focal adhesion kinase axis sensitizes breast cancer cells to ErbB2 antagonists. Cancer Res. 2010, 70, 2256–2263. [Google Scholar] [CrossRef]
- Huafeng, J.; Deqing, Z.; Yong, D.; Yulian, Z.; Ailing, H. A cross-talk between integrin β4 and epidermal growth factor receptor induces gefitinib chemoresistance to gastric cancer. Cancer Cell Int. 2018, 18. [Google Scholar] [CrossRef]
- Giannelli, G.; Azzariti, A.; Fransvea, E.; Porcelli, L.; Antonaci, S.; Paradiso, A. Laminin-5 offsets the efficacy of gefitinib (‘Iressa’) in hepatocellular carcinoma cells. Br. J. Cancer 2004, 91, 1964–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, G.A.; Xiao, B.; Zhao, H.; Al-Zahrani, K.N.; Hasim, M.S.; Villeneuve, J.; Sekhon, H.S.; Goss, G.D.; Sabourin, L.A.; Dimitroulakos, J.; et al. Focal adhesion kinase inhibitors in combination with erlotinib demonstrate enhanced anti-tumor activity in non-small cell lung cancer. PLoS ONE 2016, 11, e0150567. [Google Scholar] [CrossRef]
- Ichihara, E.; Westover, D.; Meador, C.B.; Yan, Y.; Bauer, J.A.; Lu, P.; Ye, F.; Kulick, A.; de Stanchina, E.; McEwen, R.; et al. SFK/FAK signaling attenuates osimertinib efficacy in both drug-sensitive and drug-resistant models of EGFR-mutant lung cancer. Cancer Res. 2017, 77, 2990–3000. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Sonoda, K.; Abe, H.; Watari, K.; Kusakabe, D.; Azuma, K.; Kawahara, A.; Akiba, J.; Oneyama, C.; Pachter, J.A.; et al. The activation of SRC family kinases and focal adhesion kinase with the loss of the amplified, mutated EGFR gene contributes to the resistance to afatinib, erlotinib and osimertinib in human lung cancer cells. Oncotarget 2017, 8, 70736–70751. [Google Scholar] [CrossRef] [Green Version]
- Solanki, H.S.; Raja, R.; Zhavoronkov, A.; Ozerov, I.V.; Artemov, A.V.; Advani, J.; Radhakrishnan, A.; Babu, N.; Puttamallesh, V.N.; Syed, N.; et al. Targeting focal adhesion kinase overcomes erlotinib resistance in smoke induced lung cancer by altering phosphorylation of epidermal growth factor receptor. Oncoscience 2018, 5, 21–38. [Google Scholar] [Green Version]
- Barkan, D.; Chambers, A.F. β1-Integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef]
- Cordes, D.N.; Park, C.C. beta1 integrin as a molecular therapeutic target. Int. J. Radiat. Biol. 2007, 83, 753–760. [Google Scholar] [CrossRef]
- Schaffner, F.; Ray, A.M.; Dontenwill, M. Integrin α5β1, the fibronectin receptor, as a pertinent therapeutic target in solid tumors. Cancers 2013, 5, 27–47. [Google Scholar] [CrossRef]
- Blandin, A.-F.; Renner, G.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. β1 integrins as therapeutic targets to disrupt hallmarks of cancer. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef]
- Chen, M.B.; Lamar, J.M.; Li, R.; Hynes, R.O.; Kamm, R.D. Elucidation of the roles of tumor integrin β1 in the extravasation stage of the metastasis cascade. Cancer Res. 2016, 76, 2513–2524. [Google Scholar] [CrossRef] [Green Version]
- Lahlou, H.; Muller, W.J. β1-integrins signaling and mammary tumor progression in transgenic mouse models: Implications for human breast cancer. Breast Cancer Res. 2011, 13, 229. [Google Scholar] [CrossRef]
- Morello, V.; Cabodi, S.; Sigismund, S.; Camacho-Leal, M.P.; Repetto, D.; Volante, M.; Papotti, M.; Turco, E.; Defilippi, P. [beta]1 integrin controls EGFR signaling and tumorigenic properties of lung cancer cells. Oncogene 2011, 30, 4087–4096. [Google Scholar] [CrossRef]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U.; Muller, W.J. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef]
- Aoudjit, F.; Vuori, K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 2001, 20, 4995–5004. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, T.N.; Burger, J.A.; Glodek, A.; Fujii, N.; Burger, M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene 2005, 24, 4462–4471. [Google Scholar] [CrossRef] [Green Version]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef]
- Janouskova, H.; Ray, A.-M.; Noulet, F.; Lelong-Rebel, I.; Choulier, L.; Schaffner, F.; Lehmann, M.; Martin, S.; Teisinger, J.; Dontenwill, M. Activation of p53 pathway by Nutlin-3a inhibits the expression of the therapeutic target α5 integrin in colon cancer cells. Cancer Lett. 2013, 336, 307–318. [Google Scholar] [CrossRef]
- Klobučar, M.; Grbčić, P.; Pavelić, S.K.; Jonjić, N.; Visentin, S.; Sedić, M. Acid ceramidase inhibition sensitizes human colon cancer cells to oxaliplatin through downregulation of transglutaminase 2 and β1 integrin/FAK-mediated signalling. Biochem. Biophys. Res. Commun. 2018, 503, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Maglott, A.; Bartik, P.; Cosgun, S.; Klotz, P.; Rondé, P.; Fuhrmann, G.; Takeda, K.; Martin, S.; Dontenwill, M. The small alpha5beta1 integrin antagonist, SJ749, reduces proliferation and clonogenicity of human astrocytoma cells. Cancer Res. 2006, 66, 6002–6007. [Google Scholar] [CrossRef]
- Naci, D.; El Azreq, M.-A.; Chetoui, N.; Lauden, L.; Sigaux, F.; Charron, D.; Al-Daccak, R.; Aoudjit, F. α2β1 integrin promotes chemoresistance against doxorubicin in cancer cells through extracellular signal-regulated kinase (ERK). J. Biol. Chem. 2012, 287, 17065–17076. [Google Scholar] [CrossRef] [PubMed]
- Naci, D.; Vuori, K.; Aoudjit, F. Alpha2beta1 integrin in cancer development and chemoresistance. Semin. Cancer Biol. 2015, 35, 145–153. [Google Scholar] [CrossRef]
- Pontiggia, O.; Sampayo, R.; Raffo, D.; Motter, A.; Xu, R.; Bissell, M.J.; de Kier Joffé, E.B.; Simian, M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res. Treat. 2012, 133, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Renner, G.; Janouskova, H.; Noulet, F.; Koenig, V.; Guerin, E.; Bär, S.; Nuesch, J.; Rechenmacher, F.; Neubauer, S.; Kessler, H.; et al. Integrin α5β1 and p53 convergent pathways in the control of anti-apoptotic proteins PEA-15 and survivin in high-grade glioma. Cell Death Differ. 2016, 23, 640–653. [Google Scholar] [CrossRef]
- Yang, D.; Shi, J.; Fu, H.; Wei, Z.; Xu, J.; Hu, Z.; Zhang, Y.; Yan, R.; Cai, Q. Integrinβ1 modulates tumour resistance to gemcitabine and serves as an independent prognostic factor in pancreatic adenocarcinomas. Tumour Biol. 2016, 37, 12315–12327. [Google Scholar] [CrossRef]
- Dickreuter, E.; Eke, I.; Krause, M.; Borgmann, K.; van Vugt, M.A.; Cordes, N. Targeting of β1 integrins impairs DNA repair for radiosensitization of head and neck cancer cells. Oncogene 2016, 35, 1353–1362. [Google Scholar] [CrossRef]
- Eke, I.; Dickreuter, E.; Cordes, N. Enhanced radiosensitivity of head and neck squamous cell carcinoma cells by β1 integrin inhibition. Radiother. Oncol. 2012, 104, 235–242. [Google Scholar] [CrossRef]
- Eke, I.; Zscheppang, K.; Dickreuter, E.; Hickmann, L.; Mazzeo, E.; Unger, K.; Krause, M.; Cordes, N. Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. JNCI J. Natl. Cancer Inst. 2015, 107, dju419. [Google Scholar] [CrossRef]
- Koppenhagen, P.; Dickreuter, E.; Cordes, N. Head and neck cancer cell radiosensitization upon dual targeting of c-Abl and beta1-integrin. Radiother. Oncol. 2017, 124, 370–378. [Google Scholar] [CrossRef]
- Ahmed, K.M.; Zhang, H.; Park, C.C. NF-κB regulates radioresistance mediated by β1-integrin in three-dimensional culture of breast cancer cells. Cancer Res. 2013, 73, 3737–3748. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.-M.; Onodera, Y.; Bissell, M.J.; Park, C.C. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 2010, 70, 5238–5248. [Google Scholar] [CrossRef]
- Dong, X.; Luo, Z.; Liu, T.; Chai, J.; Ke, Q.; Shen, L. Identification of integrin β1 as a novel PAG1-interacting protein involved in the inherent radioresistance of human laryngeal carcinoma. J. Cancer 2018, 9, 4128–4138. [Google Scholar] [CrossRef]
- Li, L.; Dong, X.; Peng, F.; Shen, L. Integrin β1 regulates the invasion and radioresistance of laryngeal cancer cells by targeting CD147. Cancer Cell Int. 2018, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Petrás, M.; Lajtos, T.; Friedländer, E.; Klekner, A.; Pintye, E.; Feuerstein, B.G.; Szöllosi, J.; Vereb, G. Molecular interactions of ErbB1 (EGFR) and integrin-β1 in astrocytoma frozen sections predict clinical outcome and correlate with Akt-mediated in vitro radioresistance. Neuro Oncol. 2013, 15, 1027–1040. [Google Scholar] [CrossRef] [Green Version]
- Vehlow, A.; Klapproth, E.; Storch, K.; Dickreuter, E.; Seifert, M.; Dietrich, A.; Bütof, R.; Temme, A.; Cordes, N.; Vehlow, A.; et al. Adhesion- and stress-related adaptation of glioma radiochemoresistance is circumvented by β1 integrin/JNK co-targeting. Oncotarget 2017, 8, 49224–49237. [Google Scholar] [CrossRef] [Green Version]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef]
- Miyamoto, S.; Teramoto, H.; Gutkind, J.S.; Yamada, K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: Roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996, 135, 1633–1642. [Google Scholar] [CrossRef]
- Al-Akhrass, H.; Naves, T.; Vincent, F.; Magnaudeix, A.; Durand, K.; Bertin, F.; Melloni, B.; Jauberteau, M.-O.; Lalloué, F. Sortilin limits EGFR signaling by promoting its internalization in lung cancer. Nat. Commun. 2017, 8, 1182. [Google Scholar] [CrossRef] [Green Version]
- Caswell, P.T.; Chan, M.; Lindsay, A.J.; McCaffrey, M.W.; Boettiger, D.; Norman, J.C. Rab-coupling protein coordinates recycling of α5β1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 2008, 183, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Hang, Q.; Isaji, T.; Hou, S.; Im, S.; Fukuda, T.; Gu, J. Integrin α5 suppresses the phosphorylation of epidermal growth factor receptor and its cellular signaling of cell proliferation via N-glycosylation. J. Biol. Chem. 2015, 290, 29345–29360. [Google Scholar] [CrossRef] [PubMed]
- Mattila, E.; Pellinen, T.; Nevo, J.; Vuoriluoto, K.; Arjonen, A.; Ivaska, J. Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 2005, 7, 78–85. [Google Scholar] [CrossRef]
- Cabodi, S.; Morello, V.; Masi, A.; Cicchi, R.; Broggio, C.; Distefano, P.; Brunelli, E.; Silengo, L.; Pavone, F.; Arcangeli, A.; et al. Convergence of integrins and EGF receptor signaling via PI3K/Akt/FoxO pathway in early gene Egr-1 expression. J. Cell. Physiol. 2009, 218, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.-M.; Fazekas, Z.; Petrás, M.; Nagy, P.; Sebestyén, Z.; Isola, J.; Tímár, J.; Park, J.W.; Vereb, G.; Szöllősi, J. Associations of ErbB2, β1-integrin and lipid rafts on Herceptin (Trastuzumab) resistant and sensitive tumor cell lines. Cancer Lett. 2005, 227, 201–212. [Google Scholar] [CrossRef]
- Klapproth, E.; Dickreuter, E.; Zakrzewski, F.; Seifert, M.; Petzold, A.; Dahl, A.; Schröck, E.; Klink, B.; Cordes, N. Whole exome sequencing identifies mTOR and KEAP1 as potential targets for radiosensitization of HNSCC cells refractory to EGFR and β1 integrin inhibition. Oncotarget 2018, 9, 18099–18114. [Google Scholar] [CrossRef] [Green Version]
- Zscheppang, K.; Kurth, I.; Wachtel, N.; Dubrovska, A.; Kunz-Schughart, L.A.; Cordes, N. Efficacy of Beta1 integrin and EGFR targeting in sphere-forming human head and neck cancer cells. J. Cancer 2016, 7, 736. [Google Scholar] [CrossRef] [PubMed]
- Poschau, M.; Dickreuter, E.; Singh-Müller, J.; Zscheppang, K.; Eke, I.; Liersch, T.; Cordes, N. EGFR and β1-integrin targeting differentially affect colorectal carcinoma cell radiosensitivity and invasion. Radiother. Oncol. 2015, 116, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.-F.; SU, B.; ZHAO, Y.-M.; TANG, L.; ZHANG, J.; ZHOU, C.-C. Integrin β1-mediated acquired gefitinib resistance in non-small cell lung cancer cells occurs via the phosphoinositide 3-kinase-dependent pathway. Oncol. Lett. 2016, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Zhou, C.; Li, W.; Yan, L. Integrin beta1 over-expression associates with resistance to tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. J. Cell. Biochem. 2010, 111, 1565–1574. [Google Scholar] [CrossRef]
- Ju, L.; Zhou, C. Association of integrin beta1 and c-MET in mediating EGFR TKI gefitinib resistance in non-small cell lung cancer. Cancer Cell Int. 2013, 13, 15. [Google Scholar] [CrossRef]
- Mousson, A.; Sick, E.; Carl, P.; Dujardin, D.; De Mey, J.; Rondé, P. Targeting focal adhesion kinase using inhibitors of protein-protein interactions. Cancers 2018, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Nixon, N.A.; Hannouf, M.B.; Verma, S. A review of the value of human epidermal growth factor receptor 2 (HER2)-targeted therapies in breast cancer. Eur. J. Cancer 2018, 89, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Huck, L.; Pontier, S.M.; Zuo, D.M.; Muller, W.J. β1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 15559–15564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesniak, D.; Xu, Y.; Deschenes, J.; Lai, R.; Thoms, J.; Murray, D.; Gosh, S.; Mackey, J.R.; Sabri, S.; Abdulkarim, B. Beta1-integrin circumvents the antiproliferative effects of trastuzumab in human epidermal growth factor receptor-2-positive breast cancer. Cancer Res. 2009, 69, 8620–8628. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.R.; Zhang, H.; Ziaee, S.; Ruiz-Saenz, A.; Gulizia, N.; Oeffinger, J.; Amin, D.N.; Ahuja, D.; Moasser, M.M.; Park, C.C. Effective treatment of HER2-amplified breast cancer by targeting HER3 and β1 integrin. Breast Cancer Res. Treat. 2016, 155, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Endo, Y.; Shen, Y.; Youssef, L.A.; Mohan, N.; Wu, W.J. T-DM1-resistant cells gain high invasive activity via EGFR and integrin cooperated pathways. mAbs 2018, 10, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Sauveur, J.; Matera, E.-L.; Chettab, K.; Valet, P.; Guitton, J.; Savina, A.; Dumontet, C. Esophageal cancer cells resistant to T-DM1 display alterations in cell adhesion and the prostaglandin pathway. Oncotarget 2018, 9, 21141–21155. [Google Scholar] [PubMed] [Green Version]
- Mahdi, A.; Darvishi, B.; Majidzadeh-A, K.; Salehi, M.; Farahmand, L. Challenges facing antiangiogenesis therapy: The significant role of hypoxia-inducible factor and MET in development of resistance to anti-vascular endothelial growth factor-targeted therapies. J. Cell Physiol. 2019, 234, 5655–5663. [Google Scholar] [CrossRef]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangiri, A.; Aghi, M.K.; Carbonell, W.S. β1 Integrin: Critical path to antiangiogenic therapy resistance and beyond. Cancer Res. 2014, 74, 3–7. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.-P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- DeLay, B.M.; Jahangiri, A.; Carbonell, W.S.; Hu, Y.-L.; Tsao, S.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Aghi, M.K. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to anti-angiogenic therapy. Clin. Cancer Res. 2012, 18, 2930–2942. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Lee, J.H.; Simmons, B.H.; Wong, A.; Esparza, C.O.; Plumlee, P.A.; Feng, J.; Stewart, A.E.; Hu-Lowe, D.D.; Christensen, J.G. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010, 70, 10090–10100. [Google Scholar] [CrossRef]
- Mitra, A.K.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand-independent activation of c-Met by fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef]
- Hongu, T.; Yamauchi, Y.; Funakoshi, Y.; Katagiri, N.; Ohbayashi, N.; Kanaho, Y. Pathological functions of the small GTPase Arf6 in cancer progression: Tumor angiogenesis and metastasis. Small GTPases 2016, 7, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Mai, A.; Muharram, G.; Barrow-McGee, R.; Baghirov, H.; Rantala, J.; Kermorgant, S.; Ivaska, J. Distinct c-Met activation mechanisms induce cell rounding or invasion through pathways involving integrins, RhoA and HIP1. J. Cell Sci. 2014, 127, 1938–1952. [Google Scholar] [CrossRef]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [Green Version]
- Tripolitsioti, D.; Kumar, K.S.; Neve, A.; Migliavacca, J.; Capdeville, C.; Rushing, E.J.; Ma, M.; Kijima, N.; Sharma, A.; Pruschy, M.; et al. MAP4K4 controlled integrin β1 activation and c-Met endocytosis are associated with invasive behavior of medulloblastoma cells. Oncotarget 2018, 9, 23220–23236. [Google Scholar] [CrossRef] [Green Version]
- Barrow-McGee, R.; Kishi, N.; Joffre, C.; Ménard, L.; Hervieu, A.; Bakhouche, B.A.; Noval, A.J.; Mai, A.; Guzmán, C.; Robbez-Masson, L.; et al. Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat. Commun. 2016, 7, 11942. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. αv integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.-J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the role of RGD-recognizing integrins in cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef]
- He, J.; Wang, F.; Qi, H.; Li, Y.; Liang, H. Down-regulation of αv integrin by retroviral delivery of small interfering RNA reduces multicellular resistance of HT29. Cancer Lett. 2009, 284, 182–188. [Google Scholar] [CrossRef]
- Maubant, S.; Cruet-Hennequart, S.; Poulain, L.; Carreiras, F.; Sichel, F.; Luis, J.; Staedel, C.; Gauduchon, P. Altered adhesion properties and alpha v integrin expression in a cisplatin-resistant human ovarian carcinoma cell line. Int. J. Cancer 2002, 97, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Malric, L.; Monferran, S.; Delmas, C.; Arnauduc, F.; Dahan, P.; Boyrie, S.; Deshors, P.; Lubrano, V.; Da Mota, D.F.; Gilhodes, J.; et al. Inhibiting integrin β8 to differentiate and radiosensitize glioblastoma-initiating cells. Mol. Cancer Res. 2019, 17, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.; Brodie, C.; Finniss, S.; Berens, M.E.; Rennert, J.L.; Nelson, K.; Lemke, N.; Brown, S.L.; Hahn, D.; Neuteboom, B.; et al. Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency. Int. J. Cancer 2009, 124, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Monferran, S.; Skuli, N.; Delmas, C.; Favre, G.; Bonnet, J.; Cohen-Jonathan-Moyal, E.; Toulas, C. Alphavbeta3 and alphavbeta5 integrins control glioma cell response to ionising radiation through ILK and RhoB. Int. J. Cancer 2008, 123, 357–364. [Google Scholar] [CrossRef]
- Ning, S.; Tian, J.; Marshall, D.J.; Knox, S.J. Anti–αv integrin monoclonal antibody intetumumab enhances the efficacy of radiation therapy and reduces metastasis of human cancer xenografts in nude rats. Cancer Res. 2010, 70, 7591–7599. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Luan, W.; Deng, J.; Sa, R.; Liang, H. αV integrin induces multicellular radioresistance in human nasopharyngeal carcinoma via activating SAPK/JNK pathway. PLoS ONE 2012, 7, e38737. [Google Scholar] [CrossRef]
- Cai, W.; Chen, X. Anti-angiogenic cancer therapy based on integrin alphavbeta3 antagonism. Anticancer Agents Med. Chem. 2006, 6, 407–428. [Google Scholar] [CrossRef]
- Hsu, A.R.; Veeravagu, A.; Cai, W.; Hou, L.C.; Tse, V.; Chen, X. Integrin alpha v beta 3 antagonists for anti-angiogenic cancer treatment. Recent Pat. Anticancer Drug Discov. 2007, 2, 143–158. [Google Scholar]
- Zhang, D.; Pier, T.; McNeel, D.G.; Wilding, G.; Friedl, A. Effects of a monoclonal anti-alphavbeta3 integrin antibody on blood vessels - a pharmacodynamic study. Invest. New Drugs 2007, 25, 49–55. [Google Scholar] [CrossRef]
- Eskens, F.A.L.M.; Dumez, H.; Hoekstra, R.; Perschl, A.; Brindley, C.; Böttcher, S.; Wynendaele, W.; Drevs, J.; Verweij, J.; van Oosterom, A.T. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur. J. Cancer 2003, 39, 917–926. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.J.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.-Y.; et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef]
- Khasraw, M.; Lee, A.; McCowatt, S.; Kerestes, Z.; Buyse, M.E.; Back, M.; Kichenadasse, G.; Ackland, S.; Wheeler, H. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J. Neurooncol. 2016, 128, 163–171. [Google Scholar] [CrossRef]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Cosset, É.; Ilmjärv, S.; Dutoit, V.; Elliott, K.; von Schalscha, T.; Camargo, M.F.; Reiss, A.; Moroishi, T.; Seguin, L.; Gomez, G.; et al. Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell 2017, 32, 856–868.e5. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Barnes, L.A.; Shields, D.J.; Huang, M.; Lau, S.K.; Prévost, N.; Tarin, D.; Shattil, S.J.; Cheresh, D.A. Integrin αvβ3/c-src “Oncogenic Unit” promotes anchorage-independence and tumor progression. Nat. Med. 2009, 15, 1163–1169. [Google Scholar] [CrossRef]
- Seguin, L.; Camargo, M.F.; Wettersten, H.I.; Kato, S.; Desgrosellier, J.S.; von Schalscha, T.; Elliott, K.C.; Cosset, E.; Lesperance, J.; Weis, S.M.; et al. Galectin-3, a druggable vulnerability for KRAS-addicted cancers. Cancer Discov. 2017, 7, 1464–1479. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wojnar, J. Galectin targeted therapy in oncology: Current knowledge and perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, L.; Ma, Z.; Sun, G.; Luo, X.; Li, M.; Zhai, S.; Li, P.; Wang, X. Oncogenic miR-9 is a target of erlotinib in NSCLCs. Sci. Rep. 2015, 5, 17031. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Fan, X.; Li, W.; Ping, W.; Deng, Y.; Fu, X. miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem. Biophys. Res. Commun. 2014, 446, 179–186. [Google Scholar] [CrossRef]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef]
- Shen, H.; Zhu, F.; Liu, J.; Xu, T.; Pei, D.; Wang, R.; Qian, Y.; Li, Q.; Wang, L.; Shi, Z.; et al. Alteration in Mir-21/PTEN Expression Modulates Gefitinib Resistance in Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e103305. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Su, X.; Bai, H.; Zhao, J.; Duan, J.; An, T.; Zhuo, M.; Wang, Z.; Wu, M.; Li, Z.; et al. Identification of plasma microRNA profiles for primary resistance to EGFR-TKIs in advanced non-small cell lung cancer (NSCLC) patients with EGFR activating mutation. J. Hematol. Oncol. 2015, 8, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Yao, R.; Tang, D.; Qiu, T.; Shen, Y.; Jiao, W.; Ge, N.; Xuan, Y.; Wang, Y. Prognostic significance of microRNA expression in completely resected lung adenocarcinoma and the associated response to erlotinib. Med. Oncol. 2014, 31, 203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, Y.; Zheng, Y.; Zhang, L.; Pan, Y.; Yu, J.; Yang, M. miR-608 and miR-4513 significantly contribute to the prognosis of lung adenocarcinoma treated with EGFR-TKIs. Lab. Invest. 2019, 99, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: Results of an open-label, randomized, controlled phase II study (CERTO). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef]
- Cedra, S.; Wiegand, S.; Kolb, M.; Dietz, A.; Wichmann, G. Reduced cytokine release in ex vivo response to cilengitide and cetuximab is a marker for improved survival of head and neck cancer patients. Cancers (Basel) 2017, 9. [Google Scholar]
- Wichmann, G.; Cedra, S.; Schlegel, D.; Kolb, M.; Wiegand, S.; Boehm, A.; Hofer, M.; Dietz, A. cilengitide and cetuximab reduce cytokine production and colony formation of head and neck squamous cell carcinoma cells ex vivo. Anticancer Res. 2017, 37, 521–527. [Google Scholar] [CrossRef]
- Élez, E.; Kocáková, I.; Höhler, T.; Martens, U.M.; Bokemeyer, C.; Van Cutsem, E.; Melichar, B.; Smakal, M.; Csőszi, T.; Topuzov, E.; et al. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: The randomised phase I/II POSEIDON trial. Ann. Oncol. 2015, 26, 132–140. [Google Scholar] [CrossRef]
- Maki, R.G. Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer. J. Clin. Oncol. 2010, 28, 4985–4995. [Google Scholar] [CrossRef]
- Chen, H.X.; Sharon, E. IGF-1R as an anti-cancer target—trials and tribulations. Chin. J. Cancer 2013, 32, 242. [Google Scholar] [CrossRef]
- Saegusa, J.; Yamaji, S.; Ieguchi, K.; Wu, C.-Y.; Lam, K.S.; Liu, F.-T.; Takada, Y.K.; Takada, Y. The direct binding of insulin-like growth factor-1 (igf-1) to integrin αvβ3 is involved in igf-1 signaling. J. Biol. Chem. 2009, 284, 24106–24114. [Google Scholar] [CrossRef]
- Fujita, M.; Takada, Y.K.; Takada, Y. Insulin-like growth factor (IGF) signaling requires αvβ3-IGF1-IGF type 1 receptor (IGF1R) ternary complex formation in anchorage independence, and the complex formation does not require IGF1R and Src activation. J. Biol. Chem. 2013, 288, 3059–3069. [Google Scholar] [CrossRef]
- Fujita, M.; Ieguchi, K.; Cedano-Prieto, D.M.; Fong, A.; Wilkerson, C.; Chen, J.Q.; Wu, M.; Lo, S.-H.; Cheung, A.T.W.; Wilson, M.D.; et al. An integrin binding-defective mutant of insulin-like growth factor-1 (R36E/R37E IGF1) acts as a dominant-negative antagonist of the IGF1 receptor (IGF1R) and suppresses tumorigenesis but still binds to IGF1R. J. Biol. Chem. 2013, 288, 19593–19603. [Google Scholar] [CrossRef]
- Takada, Y.; Takada, Y.K.; Fujita, M. Crosstalk between insulin-like growth factor (IGF) receptor and integrins through direct integrin binding to IGF1. Cytokine Growth Factor Rev. 2017, 34, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasskarl, J. Sorafenib. Recent Results Cancer Res. 2010, 184, 61–70. [Google Scholar] [PubMed]
- Chong, Y.; Tang, D.; Xiong, Q.; Jiang, X.; Xu, C.; Huang, Y.; Wang, J.; Zhou, H.; Shi, Y.; Wu, X.; et al. Galectin-1 from cancer-associated fibroblasts induces epithelial-mesenchymal transition through β1 integrin-mediated upregulation of Gli1 in gastric cancer. J. Exp. Clin. Cancer Res. 2016, 35, 175. [Google Scholar] [CrossRef]
- Nam, K.; Son, S.; Oh, S.; Jeon, D.; Kim, H.; Noh, D.-Y.; Kim, S.; Shin, I. Binding of galectin-1 to integrin β1 potentiates drug resistance by promoting survivin expression in breast cancer cells. Oncotarget 2017, 8, 35804–35823. [Google Scholar] [CrossRef] [Green Version]
- He, X.-J.; Tao, H.-Q.; Hu, Z.-M.; Ma, Y.-Y.; Xu, J.; Wang, H.-J.; Xia, Y.-J.; Li, L.; Fei, B.-Y.; Li, Y.-Q.; et al. Expression of galectin-1 in carcinoma-associated fibroblasts promotes gastric cancer cell invasion through upregulation of integrin β1. Cancer Sci. 2014, 105, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Liebert, M.; Washington, R.; Wedemeyer, G.; Carey, T.E.; Grossman, H.B. Loss of co-localization of alpha 6 beta 4 integrin and collagen VII in bladder cancer. Am. J. Pathol. 1994, 144, 787–795. [Google Scholar] [PubMed]
- Rodius, S.; Indra, G.; Thibault, C.; Pfister, V.; Georges-Labouesse, E. Loss of alpha6 integrins in keratinocytes leads to an increase in TGFbeta and AP1 signaling and in expression of differentiation genes. J. Cell. Physiol. 2007, 212, 439–449. [Google Scholar] [CrossRef]
- Raymond, K.; Kreft, M.; Janssen, H.; Calafat, J.; Sonnenberg, A. Keratinocytes display normal proliferation, survival and differentiation in conditional β4-integrin knockout mice. J. Cell. Sci. 2005, 118, 1045–1060. [Google Scholar] [CrossRef]
- Faure, E.; Garrouste, F.; Parat, F.; Monferran, S.; Leloup, L.; Pommier, G.; Kovacic, H.; Lehmann, M. P2Y2 receptor inhibits EGF-induced MAPK pathway to stabilise keratinocyte hemidesmosomes. J. Cell. Sci. 2012, 125, 4264–4277. [Google Scholar] [CrossRef] [Green Version]
- Frijns, E.; Sachs, N.; Kreft, M.; Wilhelmsen, K.; Sonnenberg, A. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin {beta}4. J. Biol. Chem 2010, 285, 37650–37662. [Google Scholar] [CrossRef]
- Frijns, E.; Kuikman, I.; Litjens, S.; Raspe, M.; Jalink, K.; Ports, M.; Wilhelmsen, K.; Sonnenberg, A. Phosphorylation of threonine 1736 in the C-terminal tail of integrin β4 contributes to hemidesmosome disassembly. Mol. Biol. Cell 2012, 23, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Margadant, C.; Frijns, E.; Wilhelmsen, K.; Sonnenberg, A. Regulation of hemidesmosome disassembly by growth factor receptors. Curr. Opin. Cell Biol. 2008, 20, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Margadant, C.; van Rheenen, J.; Sonnenberg, A. Serine phosphorylation of the integrin β4 subunit is necessary for epidermal growth factor receptor–induced hemidesmosome disruption. Mol. Biol. Cell 2007, 18, 3512–3522. [Google Scholar] [CrossRef]
- Ramovs, V.; Te Molder, L.; Sonnenberg, A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017, 57–58, 213–243. [Google Scholar] [CrossRef]
- Stewart, R.L.; O’Connor, K.L. Clinical significance of the integrin α6β4 in human malignancies. Lab. Invest. 2015, 95, 976–986. [Google Scholar] [CrossRef] [Green Version]
- De Arcangelis, A.; Hamade, H.; Alpy, F.; Normand, S.; Bruyère, E.; Lefebvre, O.; Méchine-Neuville, A.; Siebert, S.; Pfister, V.; Lepage, P.; et al. Hemidesmosome integrity protects the colon against colitis and colorectal cancer. Gut 2017, 66, 1748–1760. [Google Scholar] [CrossRef]
- Laval, S.; Laklai, H.; Fanjul, M.; Pucelle, M.; Laurell, H.; Billon-Galés, A.; Le Guellec, S.; Delisle, M.-B.; Sonnenberg, A.; Susini, C.; et al. Dual roles of hemidesmosomal proteins in the pancreatic epithelium: The phosphoinositide 3-kinase decides. Oncogene 2014, 33, 1934–1944. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.T.; Babicky, M.; Jaquish, D.; French, R.; Marayuma, K.; Mose, E.; Niessen, S.; Hoover, H.; Shields, D.; Cheresh, D.; et al. The RON-receptor regulates pancreatic cancer cell migration through phosphorylation-dependent breakdown of the hemidesmosome. Int. J. Cancer 2012, 131, 1744–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A Signaling Adapter Function for α6β4 Integrin in the Control of HGF-Dependent Invasive Growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Scartozzi, M.; Giampieri, R.; Loretelli, C.; Mandolesi, A.; del Prete, M.; Biagetti, S.; Alfonsi, S.; Faloppi, L.; Bianconi, M.; Bittoni, A.; et al. Role of β4 integrin in HER-3-negative, K-RAS wild-type metastatic colorectal tumors receiving cetuximab. Future Oncol. 2013, 9, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Martins Cavaco, A.C.; Rezaei, M.; Caliandro, M.F.; Martins Lima, A.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The interaction between laminin-332 and α3β1 integrin determines differentiation and maintenance of CAFs, and supports invasion of pancreatic duct adenocarcinoma cells. Cancers (Basel) 2018, 11. [Google Scholar]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Daverey, A.; Drain, A.P.; Kidambi, S. Physical intimacy of breast cancer cells with mesenchymal stem cells elicits trastuzumab resistance through Src activation. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M.; et al. Spatial proximity to fibroblasts impacts molecular features and therapeutic sensitivity of breast cancer cells influencing clinical outcomes. Cancer Res. 2016, 76, 6495–6506. [Google Scholar] [CrossRef]
- McFarlane, S.; McFarlane, C.; Montgomery, N.; Hill, A.; Waugh, D.J.J. CD44-mediated activation of α5β1-integrin, cortactin and paxillin signaling underpins adhesion of basal-like breast cancer cells to endothelium and Fibronectin-enriched matrices. Oncotarget 2015, 6, 36762–36773. [Google Scholar] [CrossRef] [Green Version]
- Blandin, A.-F.; Noulet, F.; Renner, G.; Mercier, M.-C.; Choulier, L.; Vauchelles, R.; Ronde, P.; Carreiras, F.; Etienne-Selloum, N.; Vereb, G.; et al. Glioma cell dispersion is driven by α5 integrin-mediated cell–matrix and cell–cell interactions. Cancer Lett. 2016, 376, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 2019, 216, 688. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Guo, L.; Ren, T.; Yang, Y. Stromal extracellular matrix is a microenvironmental cue promoting resistance to EGFR tyrosine kinase inhibitors in lung cancer cells. Int. J. Biochem. Cell Biol. 2019, 106, 96–106. [Google Scholar] [CrossRef]
- Yamazaki, S.; Higuchi, Y.; Ishibashi, M.; Hashimoto, H.; Yasunaga, M.; Matsumura, Y.; Tsuchihara, K.; Tsuboi, M.; Goto, K.; Ochiai, A.; et al. Collagen type I induces EGFR-TKI resistance in EGFR-mutated cancer cells by mTOR activation through Akt-independent pathway. Cancer Sci. 2018, 109, 2063–2073. [Google Scholar] [CrossRef]
- Brighton, H.E.; Angus, S.P.; Bo, T.; Roques, J.; Tagliatela, A.C.; Darr, D.B.; Karagoz, K.; Sciaky, N.; Gatza, M.L.; Sharpless, N.E.; et al. New mechanisms of resistance to MEK inhibitors in melanoma revealed by intravital imaging. Cancer Res. 2018, 78, 542–557. [Google Scholar] [CrossRef]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Abel, E.V.; Koomen, J.M.; Fang, B.; Wood, E.R.; Chen, Y.A.; Fisher, K.J.; Iyengar, S.; Dahlman, K.B.; Wargo, J.A.; et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene 2016, 35, 1225–1235. [Google Scholar] [CrossRef]
- Margue, C.; Philippidou, D.; Kozar, I.; Cesi, G.; Felten, P.; Kulms, D.; Letellier, E.; Haan, C.; Kreis, S. Kinase inhibitor library screening identifies synergistic drug combinations effective in sensitive and resistant melanoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 56. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef]
- Elbediwy, A.; Thompson, B.J. Evolution of mechanotransduction via YAP/TAZ in animal epithelia. Current Opin. Cell Biol. 2018, 51, 117–123. [Google Scholar] [CrossRef]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.-H.; Lee, J.-K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-C.; Hsieh, T.-L.; Tiong, T.-Y.; Hsiao, C.-H.; Ji, A.T.-Q.; Hsu, W.-T.; Lee, O.K.; Ho, J.H. Regulation of metastatic ability and drug resistance in pulmonary adenocarcinoma by matrix rigidity via activating c-Met and EGFR. Biomaterials 2015, 60, 141–150. [Google Scholar] [CrossRef]
- Lin, C.-H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 2015, 26, 3946–3953. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Sleiman, M.; Moriarty, T.; Herrick, W.G.; Peyton, S.R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 2014, 35, 5749–5759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin assembly. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.S.; Park, C.J.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M.; et al. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 2016, 35, 1899–1908. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the tumor microenvironment: Shield or spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin signaling in Cancer: Mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Löffek, S.; Franzke, C.-W.; Helfrich, I. Tension in cancer. Int. J. Mol. Sci. 2016, 17, 1910. [Google Scholar] [CrossRef]
- Mercier, M.-C.; Dontenwill, M.; Choulier, L. Selection of nucleic acid aptamers targeting tumor cell-surface protein biomarkers. Cancers (Basel) 2017, 9. [Google Scholar]
- Camorani, S.; Crescenzi, E.; Gramanzini, M.; Fedele, M.; Zannetti, A.; Cerchia, L. Aptamer-mediated impairment of EGFR-integrin αvβ3 complex inhibits vasculogenic mimicry and growth of triple-negative breast cancers. Sci. Rep. 2017, 7, 46659. [Google Scholar] [CrossRef] [Green Version]
- Laurenzana, A.; Margheri, F.; Biagioni, A.; Chillà, A.; Pimpinelli, N.; Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Calorini, L.; Serratì, S.; et al. EGFR/uPAR interaction as druggable target to overcome vemurafenib acquired resistance in melanoma cells. EBioMedicine 2019, 39, 194–206. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RTK | Therapies Targeting RTK | Type of Tumor | Experimental Model | Patient Data | Integrin Modulation | Mechanisms of Resistance | Ref |
|---|---|---|---|---|---|---|---|
| β1 integrin | |||||||
| EGFR | Cetuximab | Head and neck squamous cell carcinoma | A549 cells | - | Cetuximab-induced fibronectin overexpression. siRNA-mediated depletion of β1 and α5 | Cetuximab enhances p38/ATF2-dependent fibronectin production and the activation of the focal adhesion kinase (FAK)/Erk pathway. siRNA-mediated depletion of β1 and α5 integrin decreases the cell survival of cetuximab-treated cells. | [10] |
| EGFR | Cetuximab | Pancreatic cancer | Miapaca-2, Capan-2, SW1990 AsPC-1, BXPC-3, PANC-1 | - | -Endogenous overexpression of β1 integrin in resistant cells -siRNA-mediated depletion of β1 | β1 overexpression in resistant cells stimulates Src and Akt pathways. Extracellular matrix (ECM)-independent activation of β1 is mediated by its interaction with neuropilin-1. siRNA-mediated depletion of β1 or inhibition of β1/neuropilin-1 interaction increases cetuximab cell toxicity. | [11] |
| EGFR | mAb225 | Colon cancer | Caco-2 | - | Plasmid-induced α5 overexpression | Fibronectin stimulation of α5-expressing cells overrides mAb225-mediated cell growth inhibition. Integrin activates epidermal growth factor receptor (EGFR) kinase and the mitogen-activated protein kinase (MAPK) pathway. | [12] |
| EGFR | Gefitinib Erlotinib | Lung cancer | PC-9 and 11-18 | Patient samples | -Endogenous overexpression of β1 integrin in resistant cells and tumors -siRNA-mediated depletion of β1 | siRNA-mediated silencing of β1 restores Erlotinib potency to inhibit cell proliferation and the Src and Akt pathways. | [13] |
| EGFR | PD1530335 (AG1517) | Glioma | Glioma stem-like cells (GSCs) isolated from glioblastoma (GBM) surgical pieces | - | Lentiviral-mediated β1 overexpression | Delocalization of β1 integrin from lipid raft sensitizes GSC to tyrosine kinase inhibitor (TKI)-induced apoptosis. β1 overexpression protects GSC from apoptosis in a FAK-dependent manner. | [14] |
| HER2 | Trastuzumab Lapatinib | Breast cancer | HER2+ cells (BT474, HCC1954) | - | -Endogenous overexpression of β1 integrin in resistant cells. -siRNA-mediated depletion of β1 and function-blocking mAb | Overexpression of β1 enhances FAK and Src phosphorylation. Silencing or functional inhibition of β1 integrin sensitizes cells to HER-2 inhibition (cell proliferation, apoptosis, clonogenic assays) in a FAK-dependent way. | [15] |
| HER2 | TPB (trastuzumab + pertuzumab + burparlisib) | Breast cancer | Tumors cells derived from HER2+/PIK3CAH1047R mice, MDA-MB453, HCC1954 cell lines | Patient samples and data | -Endogenous overexpression of collagen II in resistant tumors - β1 function-blocking mAb | Resistance to anti-HER2 tritherapy activates β1 integrin and Src pathways. Inhibition of β1/Src blocks coll II-induced resistance to TPB (cell growth, cell survival) | [16] |
| VEGFR | Bevacizumab | Glioma | U87, bevacizumab-resistant cell lines derived from surgical pieces (in vitro and xenografts) | Patient samples and data | -Endogenous overexpression of β1 integrin in resistant cells. -shRNA-mediated depletion of β1 and function-blocking mAb | Bevacizumab induces hypoxia that is associated with increased β1 and FAK expression. β1 inhibition (function-blocking mAb) results in increased cell apoptosis and in disrupted tumor mass formation in the treated tumor | [17] |
| VEGFR | Bevacizumab | Glioblastoma breast cancer | PDX for bevacizumab-resistant human GBM GBM and breast cancer cells | Patient samples | Increased β1/c-Met complex formation in bevacizumab-resistant tumors | Vascular endothelial growth factor receptor (VEGFR)-2 activation impedes β1/cMet complex formation. Resistance to antiangiogenic therapy increased β1/cMet complex formation and cross-activation of both receptors. | [18] |
| β 3 integrin | |||||||
| EGFR | Erlotinib Lapatinib | Lung cancer | A549 and H23 xenograft | Patient samples | shRNA-mediated depletion of β3 | EGFR TKI treatment induces selection of β3-positive cancer stem cells. Integrin β3 (in a ligand-independent way) interacts with galectin-3 to promote KRAS/RalB/NFkB activation, thereby promoting cell survival. | [19] |
| IGFR | Linsitinib | Pancreatic cancer | Panc-1 and FG xenograft | - | |||
| EGFR | Gefitinib | Lung cancer | HCC827 | - | -Epigenetic silencing of β3-targeting miR-489-3p in resistance cells -Lentivirus-mediated expression of β3 -Inhibitor or mimic of miR-489-3p | Hypermethylation of miR-483-3p in resistant cells activates the β3-dependent FAK/Erk pathway to promote cell survival and EMT | [20] |
| IGFR | Cixutumumab | Head and neck squamous cell carcinoma | Several cell lines | Patient samples | shRNA-mediated depletion of β3 and function-blocking mAb | Upon cixutumumab treatment, insulin-like growth factor (IGF)-1 directly binds to integrin ανβ3, increasing Src/Akt-dependent proliferation and survival. | [21] |
| Lung cancer | 686LN, UMNSCC38, H226B, A549 In vitro and xenograft | - | |||||
| PDGFR, VEGFR, FGFR | Sorafenib | Acute myeloid leukemia | MV4-11 | Patient samples and data | -Endogenous overexpression of β3 integrin in resistant cells - β3 function-blocking mAb | Activation of β3/PI3K/Akt/GSK3β/β-catenin pathway reduces apoptotic level and increases cell proliferation in resistant cells | [22] |
| PDGFR, VEGFR, FGFR | Sorafenib | Hepatic cancer | Huh-7, Hep3B, SK-Hep-1, HepG2, PLC/PRF/5 | - | -shRNA-mediated depletion of β3 | Forced expression of galectin-1 elevates β3 expression and activates the FAK/PI3K/Akt pathway to trigger EMT. This is correlated with an increased resistance to sorafenib in galectin-1 expressing cells. | [23] |
| β4 integrin | |||||||
| HER2 | Gefitinib | Breast cancer | Murine model mammary gland MMTV-Neu (YD) | - | -Forced expression of β4 mutant (depleted from its signaling domain) | α6β4/ErbB2 complex activates transcription factor STAT3 and c-Jun to promote cancer progression. The signaling domain of β4 is required to trigger gefitinib resistance by an unknown mechanism, whereas ErbB2, C-Jun and STAT3 phosphorylation is still inhibited by gefitinib. | [24] |
| HER2 | Trastuzumab Lapatinib | Breast cancer | BT474, ZR-75-1, SKBR3, MD-MB-453 | - | shRNA-mediated depletion of α6β4 and function-blocking mAbs | Integrin-mediated adhesion to laminin-5 promotes resistance to anti-ERB2 therapies. Removal of CD151 (an integrin co-receptor) or FAK sensitizes cells to drugs (cell proliferation) | [25] |
| EGFR | Gefitinib | Gastric cancer | SGC7901 | Patient samples | -Endogenous overexpression of α6β4 integrin in resistant cells -siRNA-mediated depletion of α6β4 | Endogenous or forced expression of β4 integrin promotes gefitinib resistance (cell proliferation and apoptosis). β4 expression is correlated with a decrease in p-EGFR protein levels. | [26] |
| EGFR | Gefitinib | Hepatic cancer | HLF, Alexander, HepG2, Sk-Hep1 | - | Laminin-332 expression | Lm-332-dependent activation of integrin dampens gefitinib effectiveness in cell proliferation survival and apoptotis assays. Lm-332 potentiates the activation of Akt in gefitinib-treated cells. | [27] |
| FAK | |||||||
| EGFR | Erlotinib | Lung cancer | A549, H1299, H1975, HCC827, HCC4006 Xenograft of A549 | - | FAK inhibitors | Combination of FAK inhibitors and erlotinib is more potent than a single agent to reduce cell viability (2D and 3D models), to increase the apoptosis pathway and cell cycle arrest in resistant cells, and to reduce tumor growth in vivo. The sensitization of erlotinib by FAK inhibitors is associated with a strong inhibition of Akt. | [28] |
| EGFR | Erlotinib Osimertinib | Lung cancer | PC-9, H1975, HCC827, HCC4006, H3255, 11-18 cell lines PC-9 xenografts | - | FAK inhibitor | Activation of FAK and Src family kinases (SFK) pathways attenuates the efficiency of EGFR therapies presumably via the sustained activation of MAPK and Akt pathways. Concomitant inhibition FAK, Src and EGFR inhibitors potently inhibit MAPK and Akt pathways and cell proliferation. | [29] |
| EGFR | Afatinib Erlotinib Osimertinib | Lung cancer | PC-9, HCC827 Established TKI-resistant cells | - | siRNA-mediated depletion of FAK and inhibitor | Compensatory activation of SFKs, FAK and Akt is observed in TKI- resistant cells. FAK inhibition increased afatinib efficacy to inhibit cell survival and cell migration. | [30] |
| EGFR | Erlotinib | Lung cancer | H1299, H1650 cell lines H358 cell line and xenograft | - | siRNA-mediated depletion of FAK and inhibitor (PF-562271) | Mass spectrometry analysis revealed an aberrant phosphorylation of FAK in erlotinib-resistant cells. Inhibition of FAK led to a decrease in cell survival in erlotinib-treated cells. | [31] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz da Silva, E.; Dontenwill, M.; Choulier, L.; Lehmann, M. Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer. Cancers 2019, 11, 692. https://doi.org/10.3390/cancers11050692
Cruz da Silva E, Dontenwill M, Choulier L, Lehmann M. Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer. Cancers. 2019; 11(5):692. https://doi.org/10.3390/cancers11050692
Chicago/Turabian StyleCruz da Silva, Elisabete, Monique Dontenwill, Laurence Choulier, and Maxime Lehmann. 2019. "Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer" Cancers 11, no. 5: 692. https://doi.org/10.3390/cancers11050692
APA StyleCruz da Silva, E., Dontenwill, M., Choulier, L., & Lehmann, M. (2019). Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer. Cancers, 11(5), 692. https://doi.org/10.3390/cancers11050692