Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines



Abstract
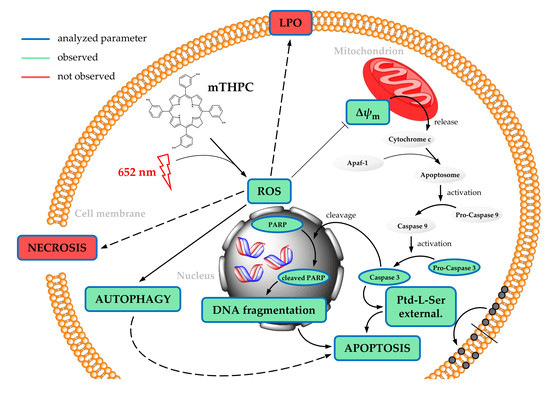
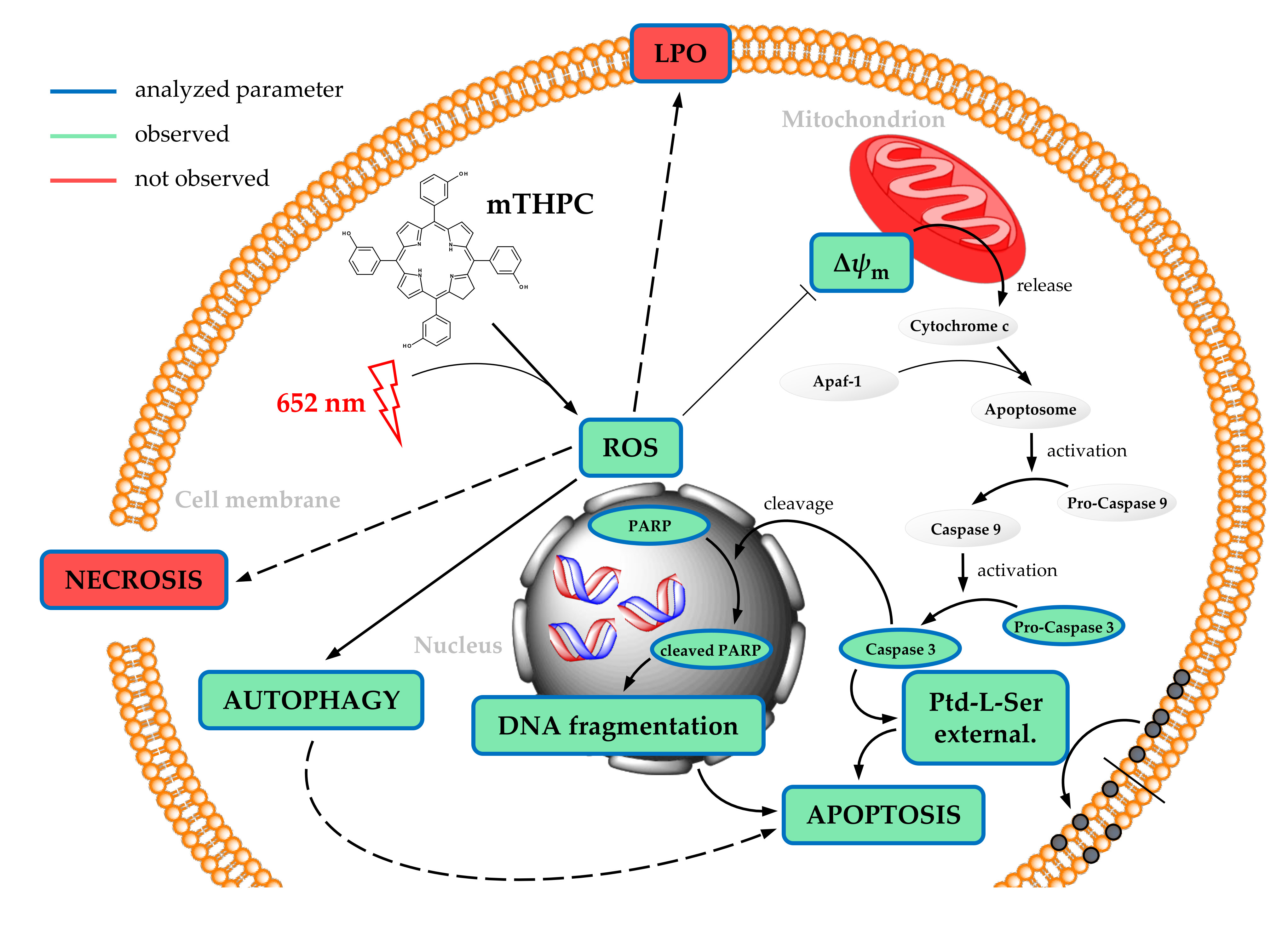
:
1. Introduction
2. Results
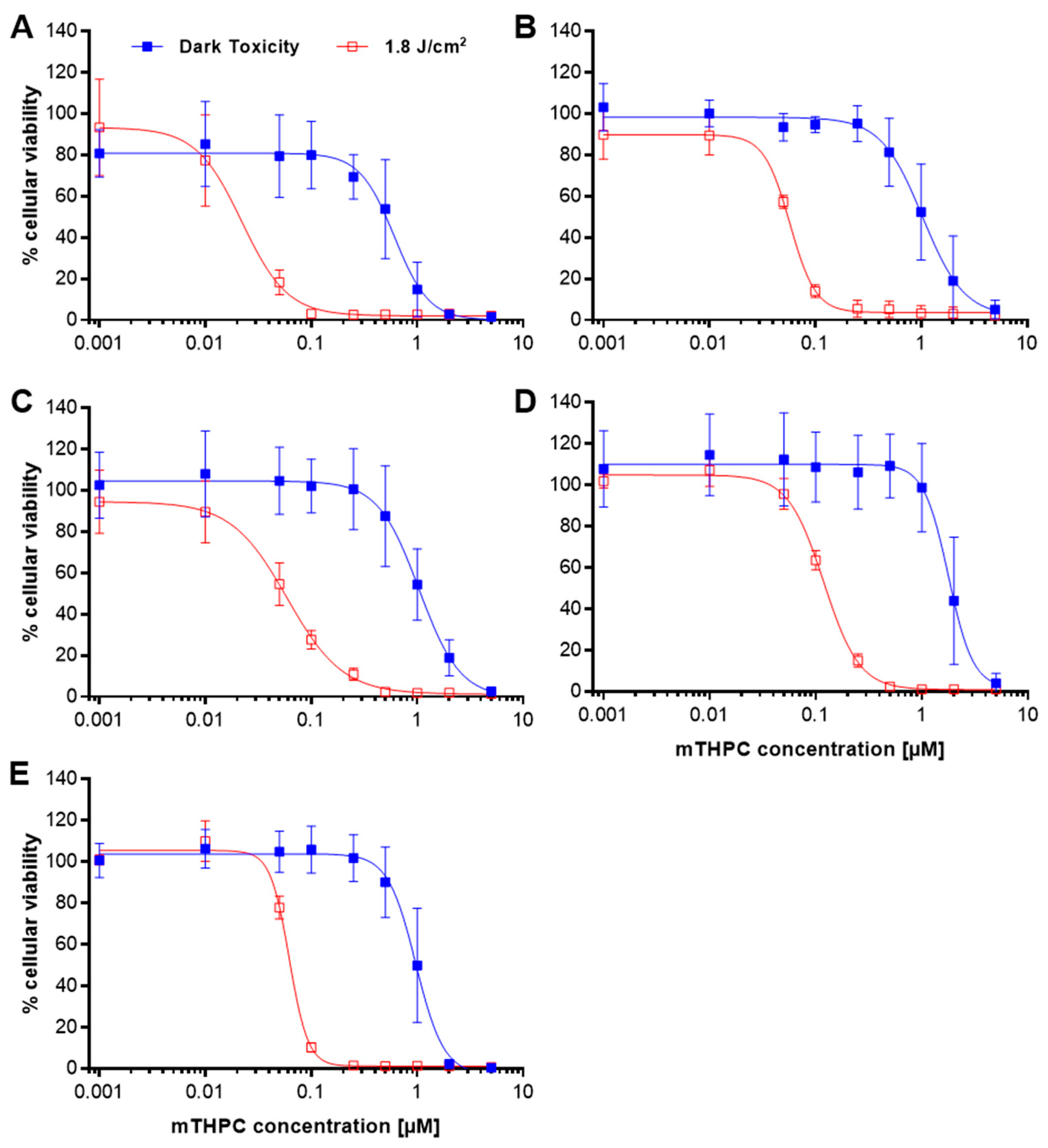
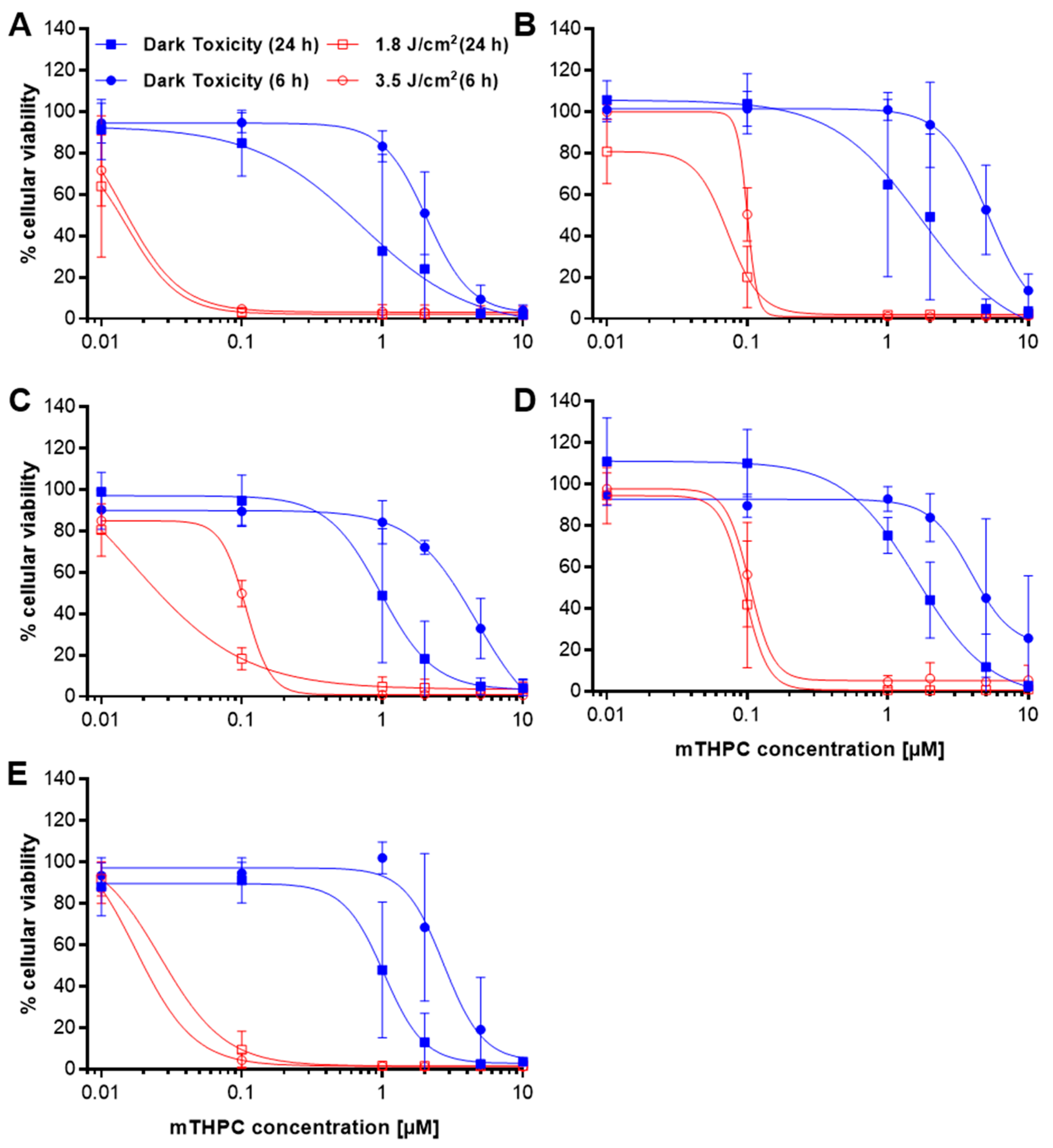
2.1. Loss of Cellular Viability after mTHPC-PDT
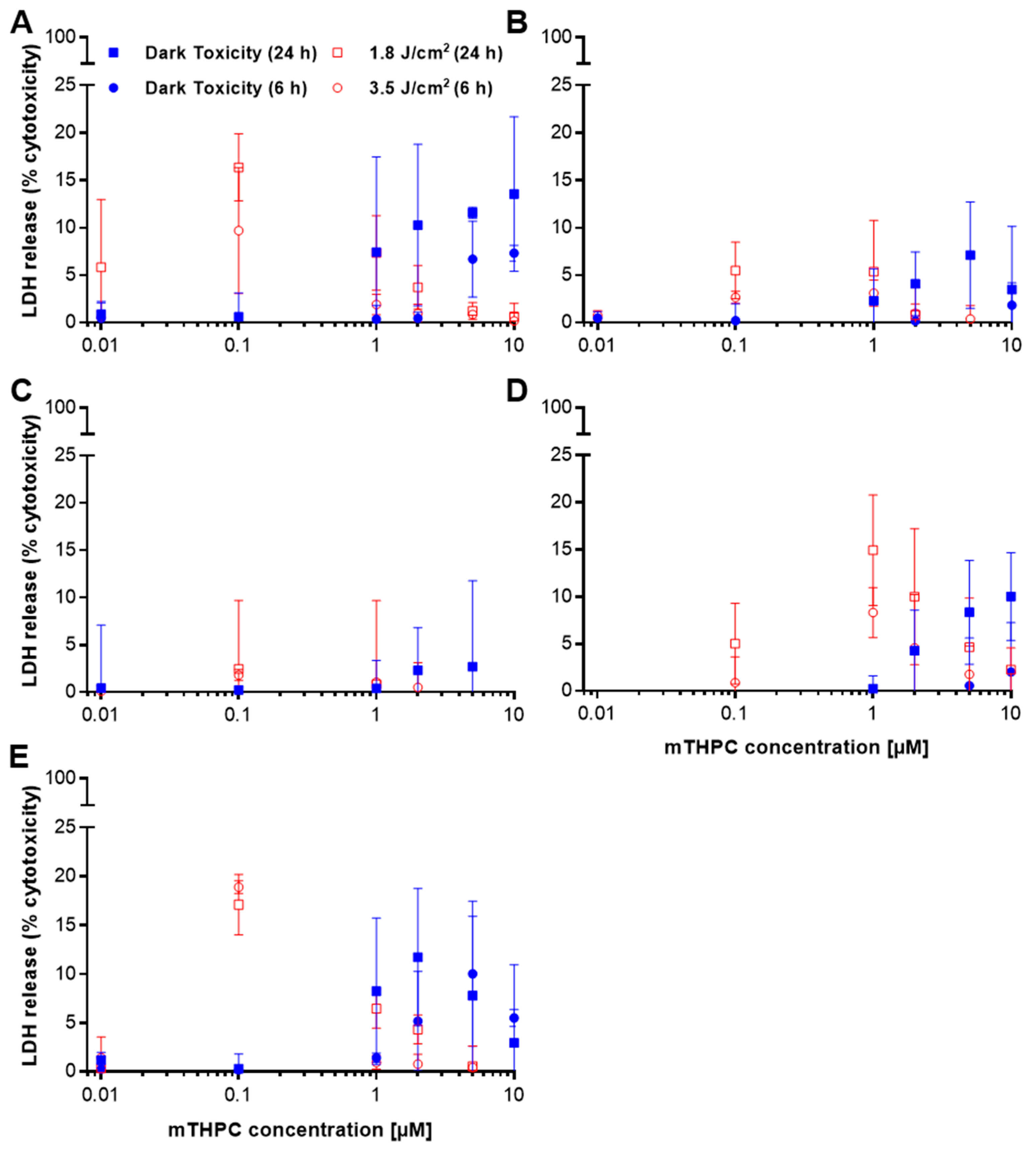
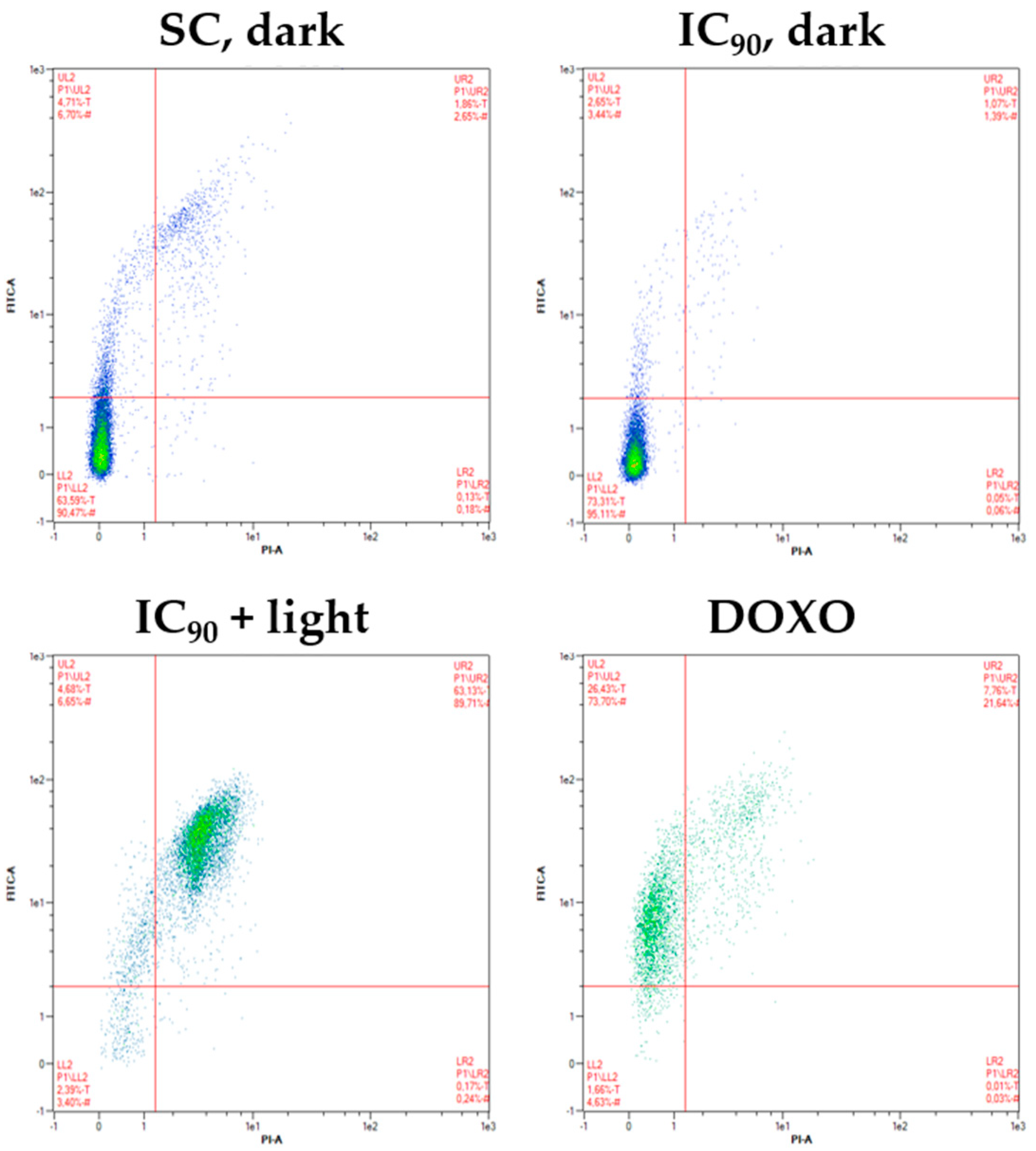
2.2. Only Minor Induction of Necrosis after mTHPC-PDT
2.3. Oxidative Stress and Apoptotis Induction After mTHPC-PDT
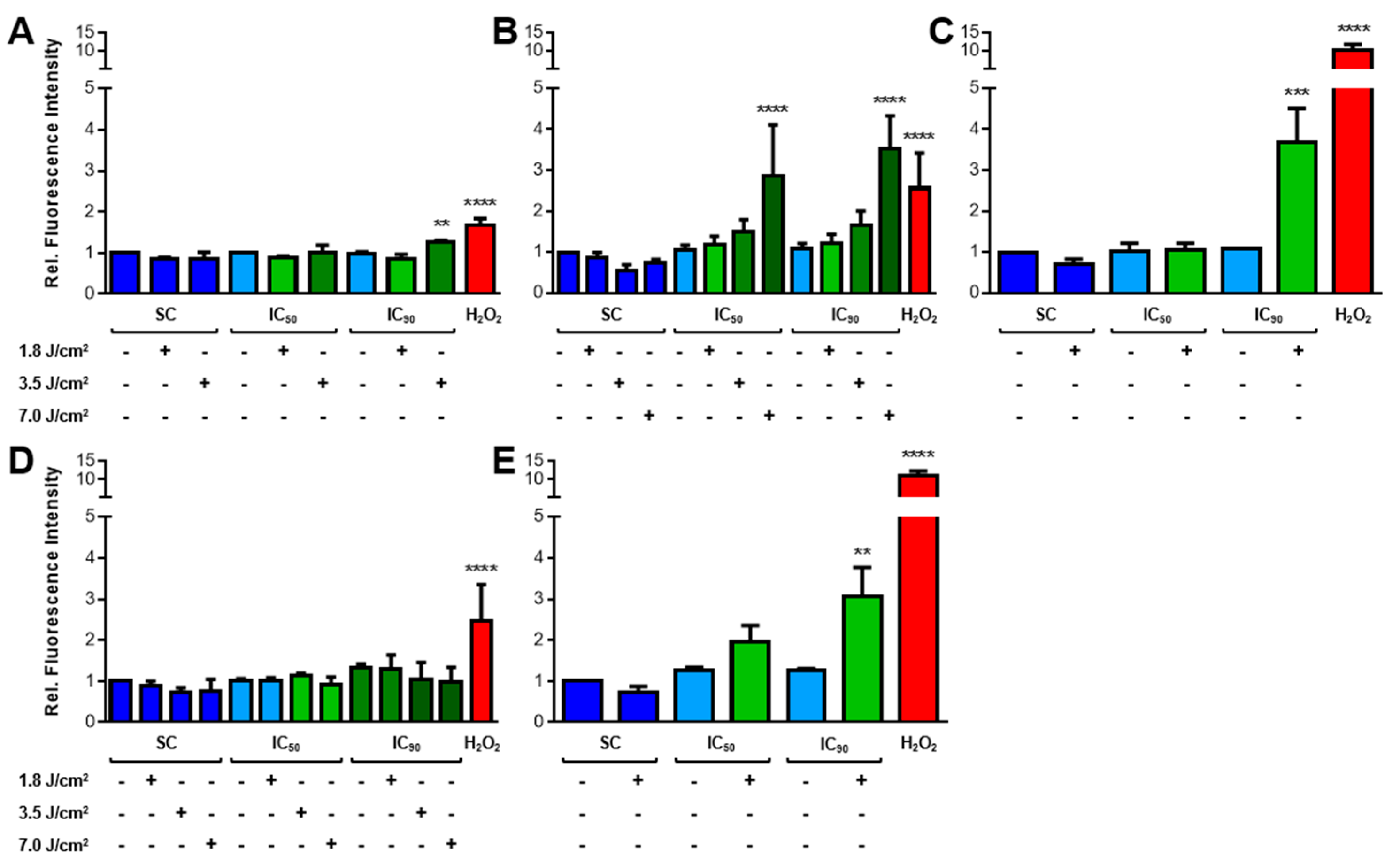
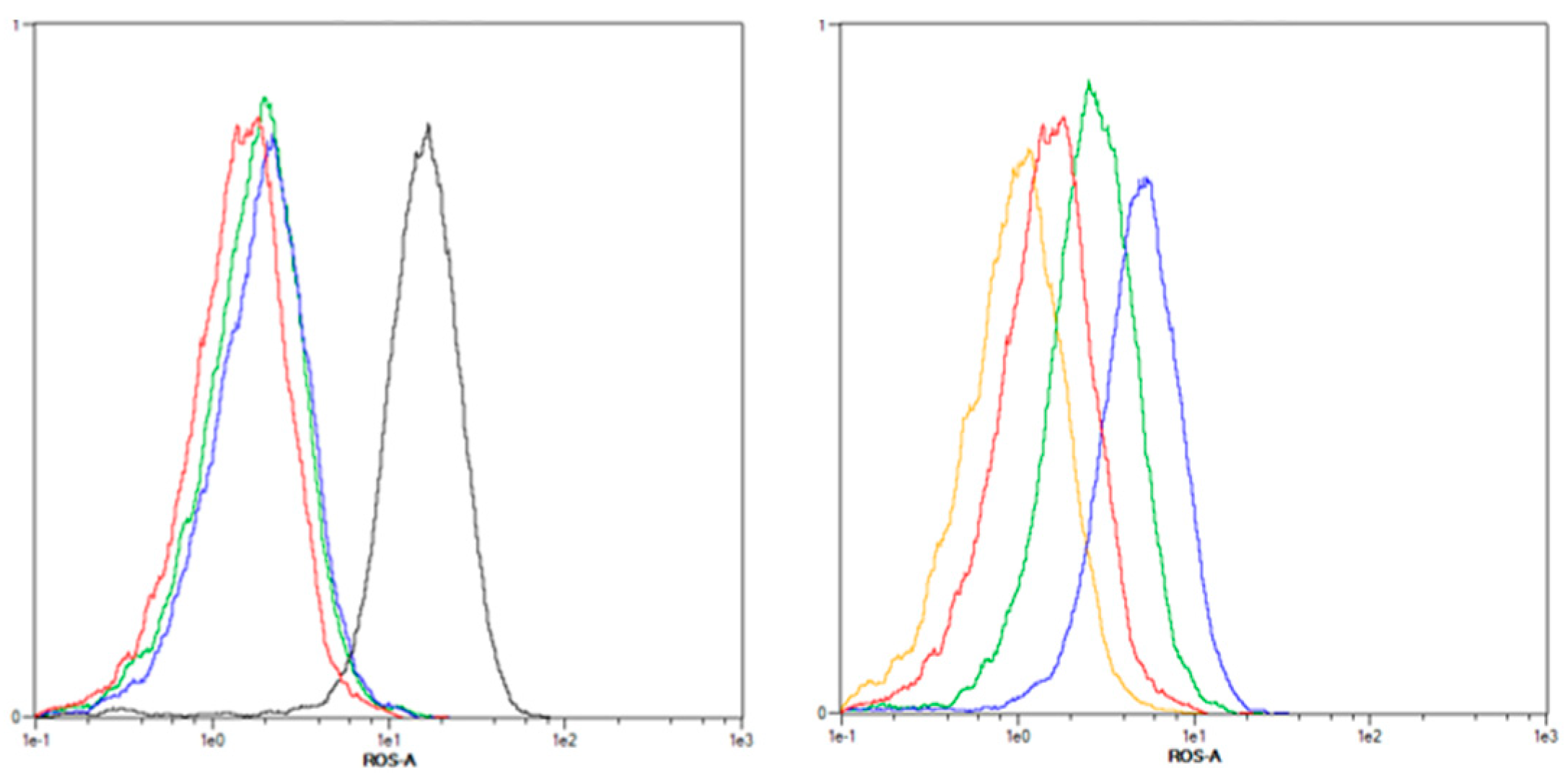
2.3.1. Increased ROS Generation after mTHPC-PDT
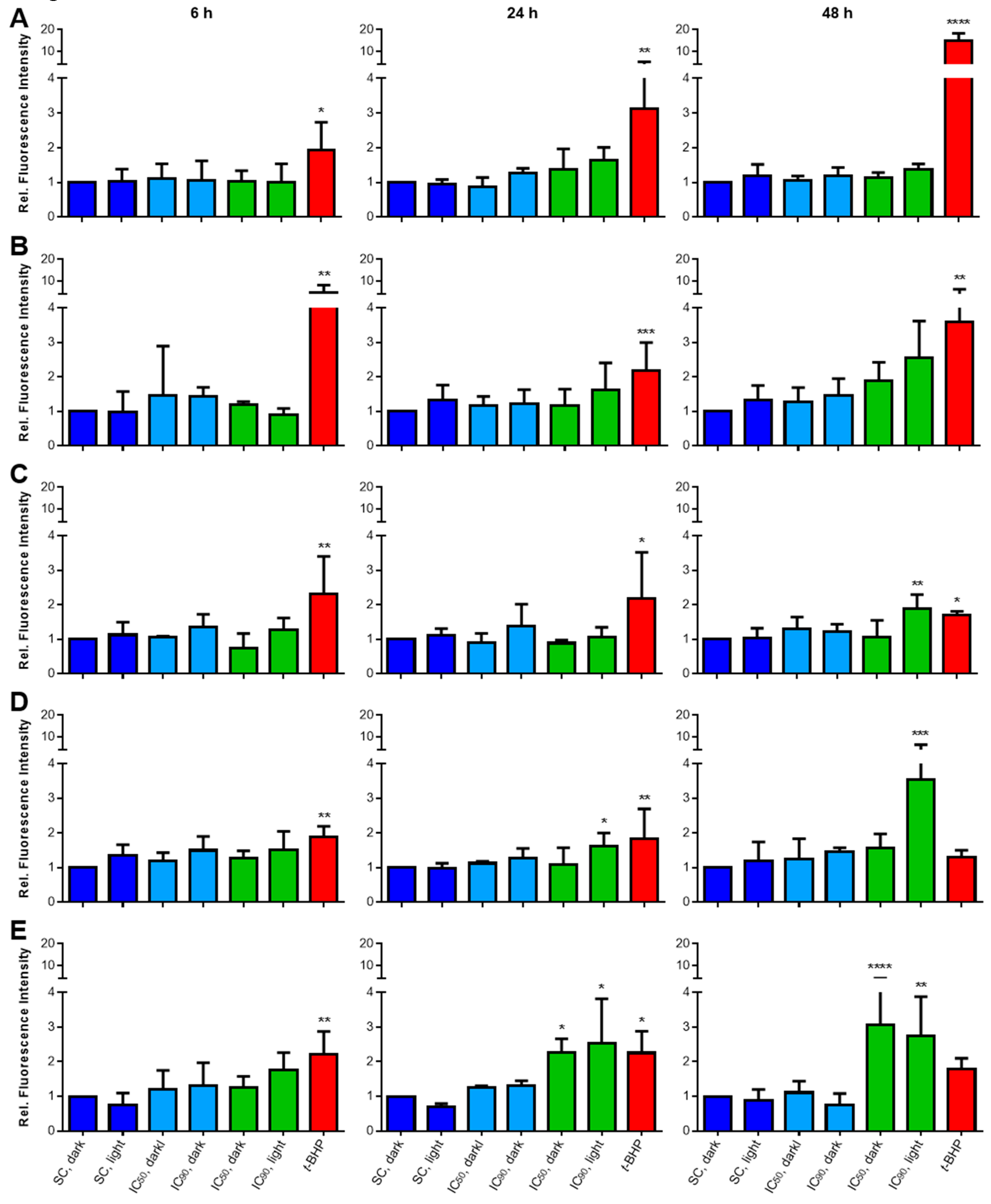
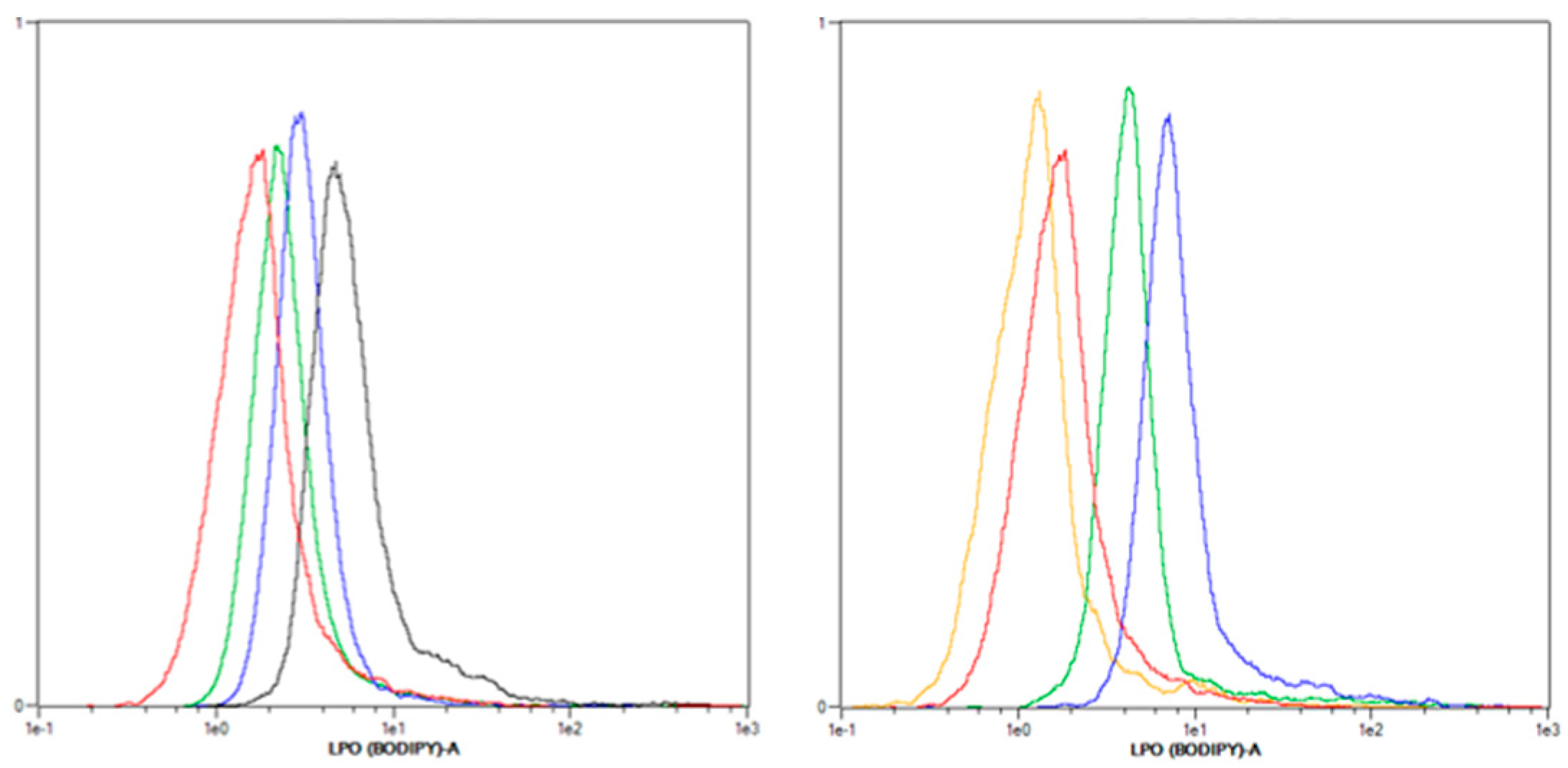
2.3.2. Lipid Peroxidation (LPO) Only Plays a Minor Role after mTHPC-PDT
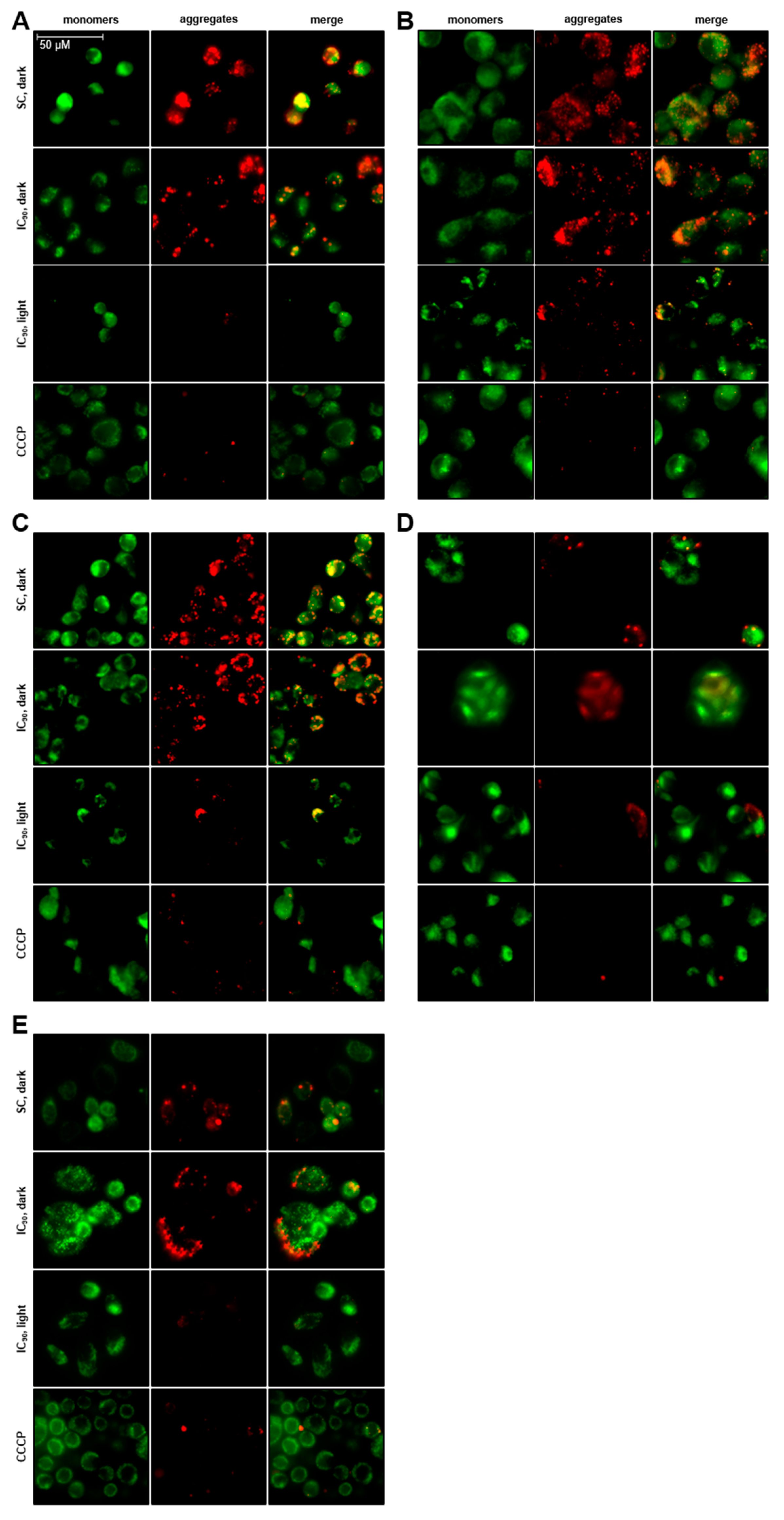
2.3.3. Total Loss of Mitochondrial Membrane Potential (δψM) after mTHPC-PDT
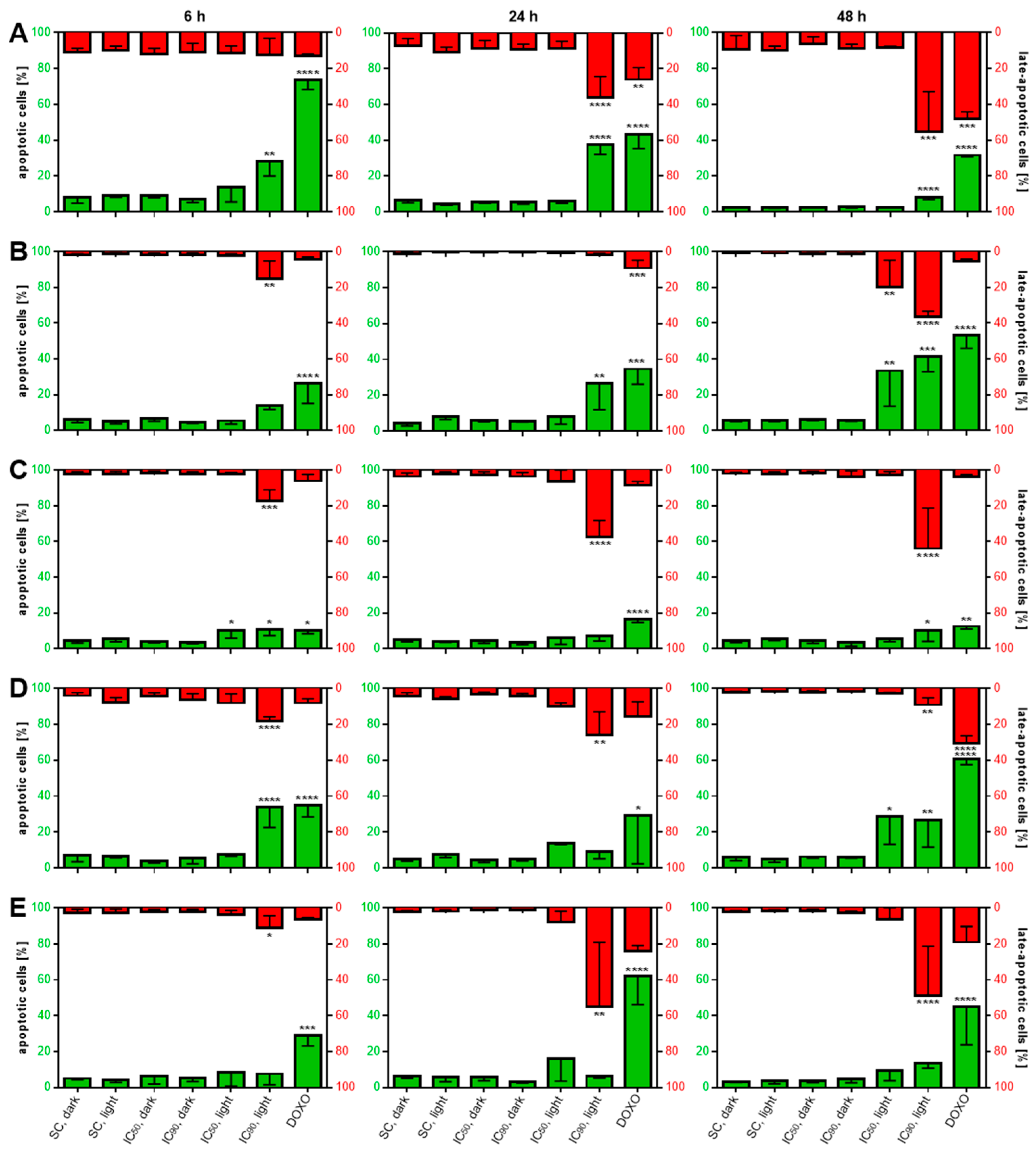
2.3.4. Induction of Phosphatidylserine Externalization after mTHPC-Mediated PDT
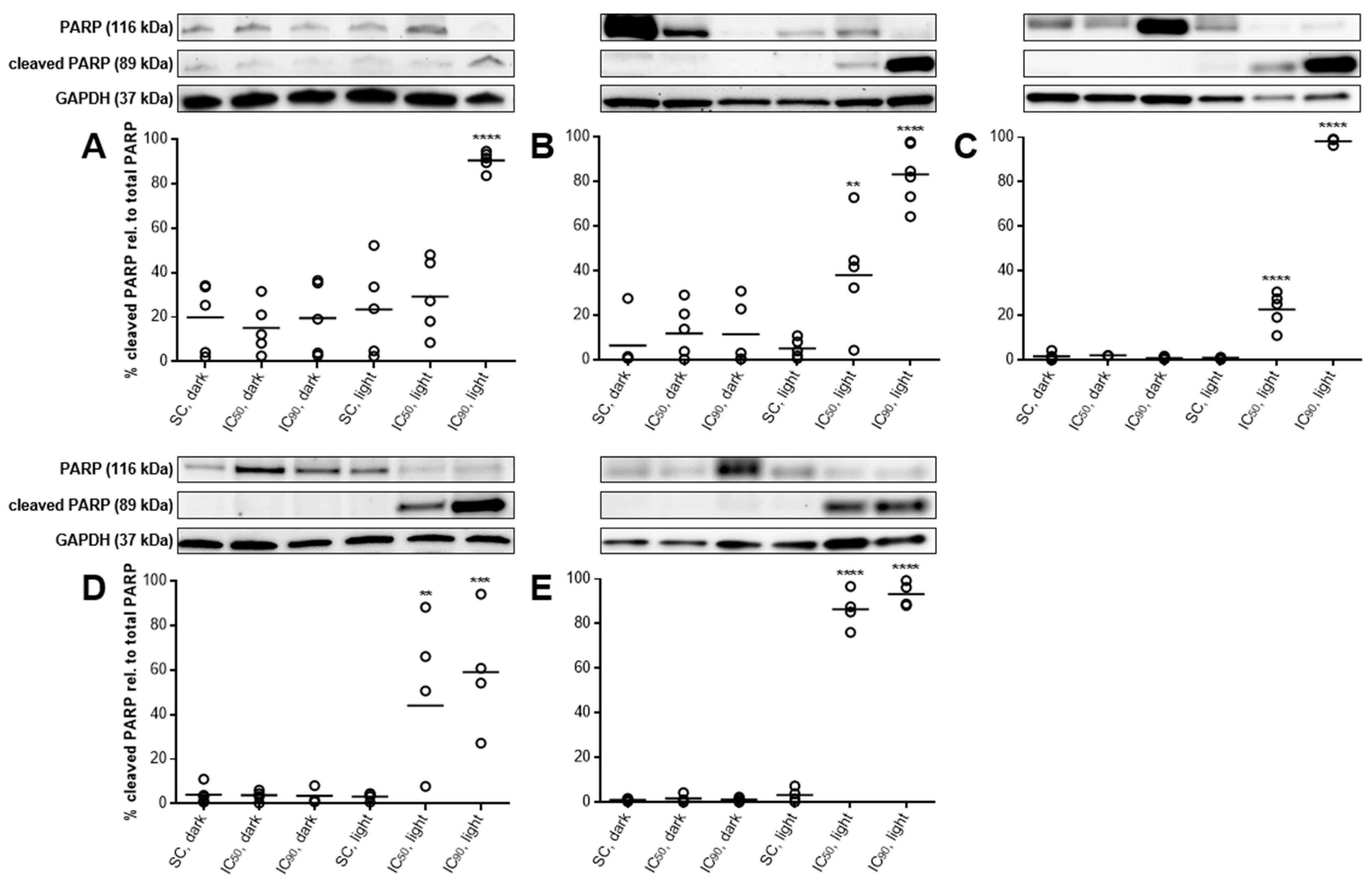
2.3.5. PARP Cleavage Confirms Induction of Apoptosis after mTHPC-PDT
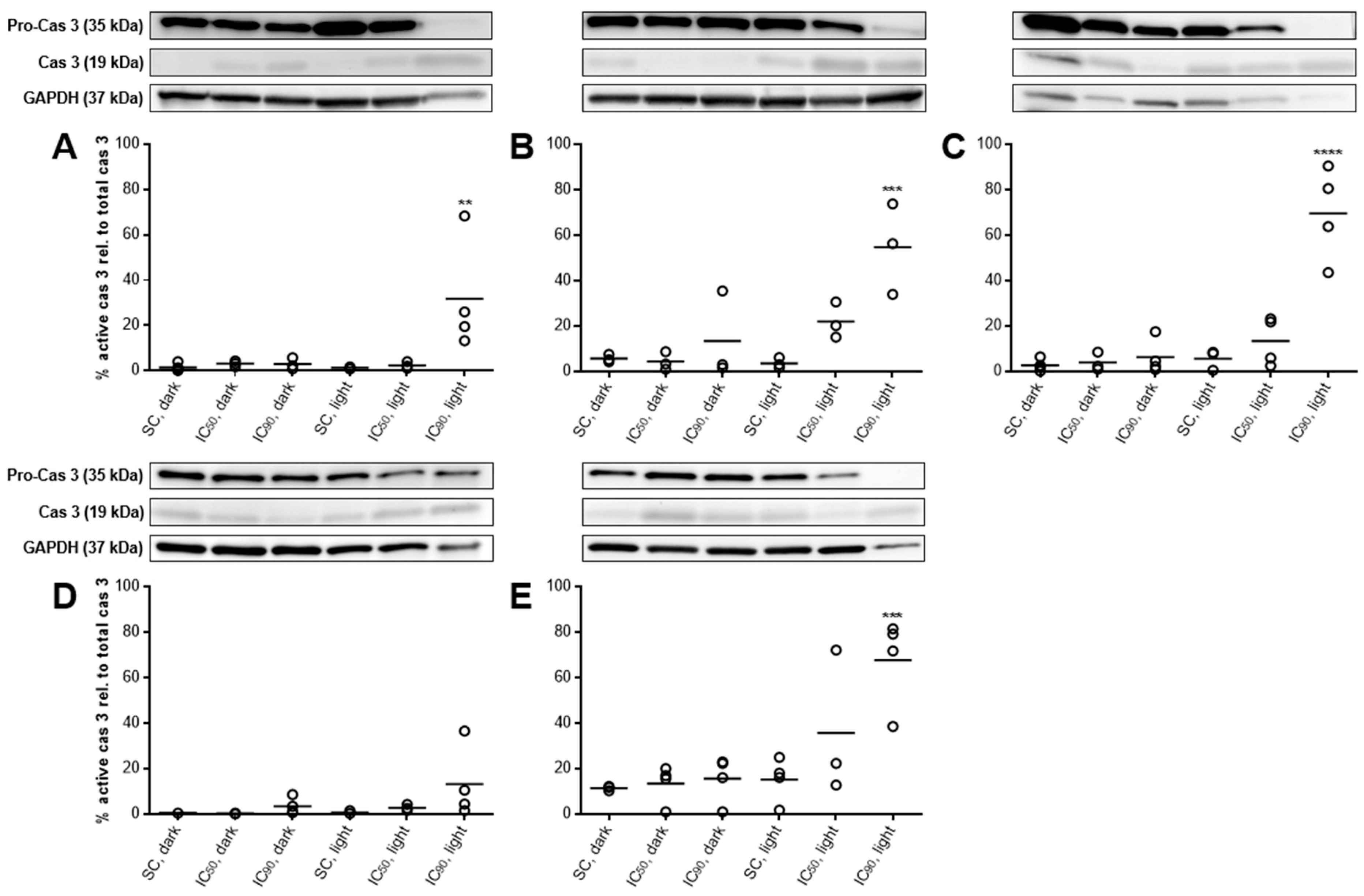
2.3.6. PARP Cleavage at Least Partly Traced Back to Caspase 3-Activation
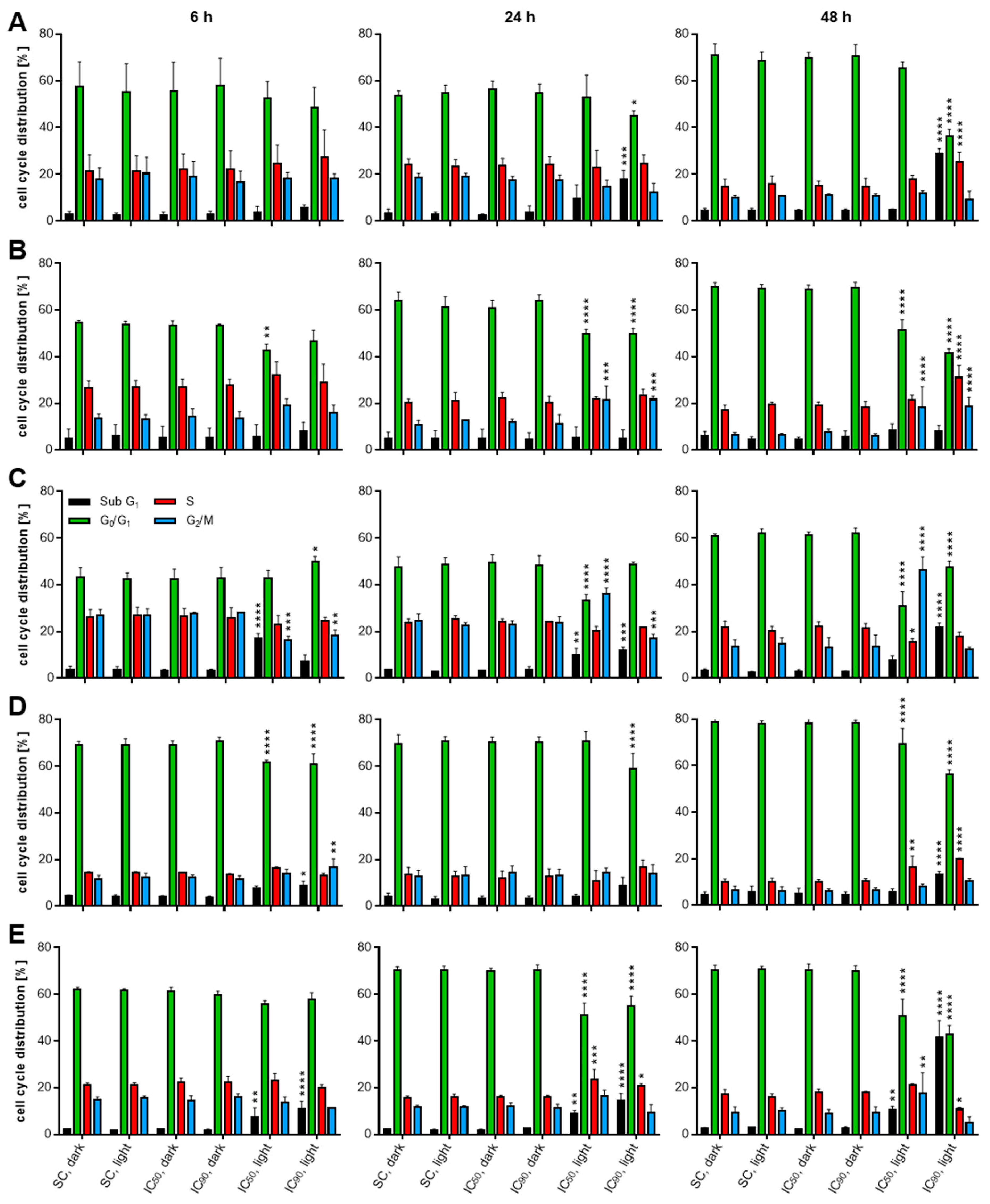
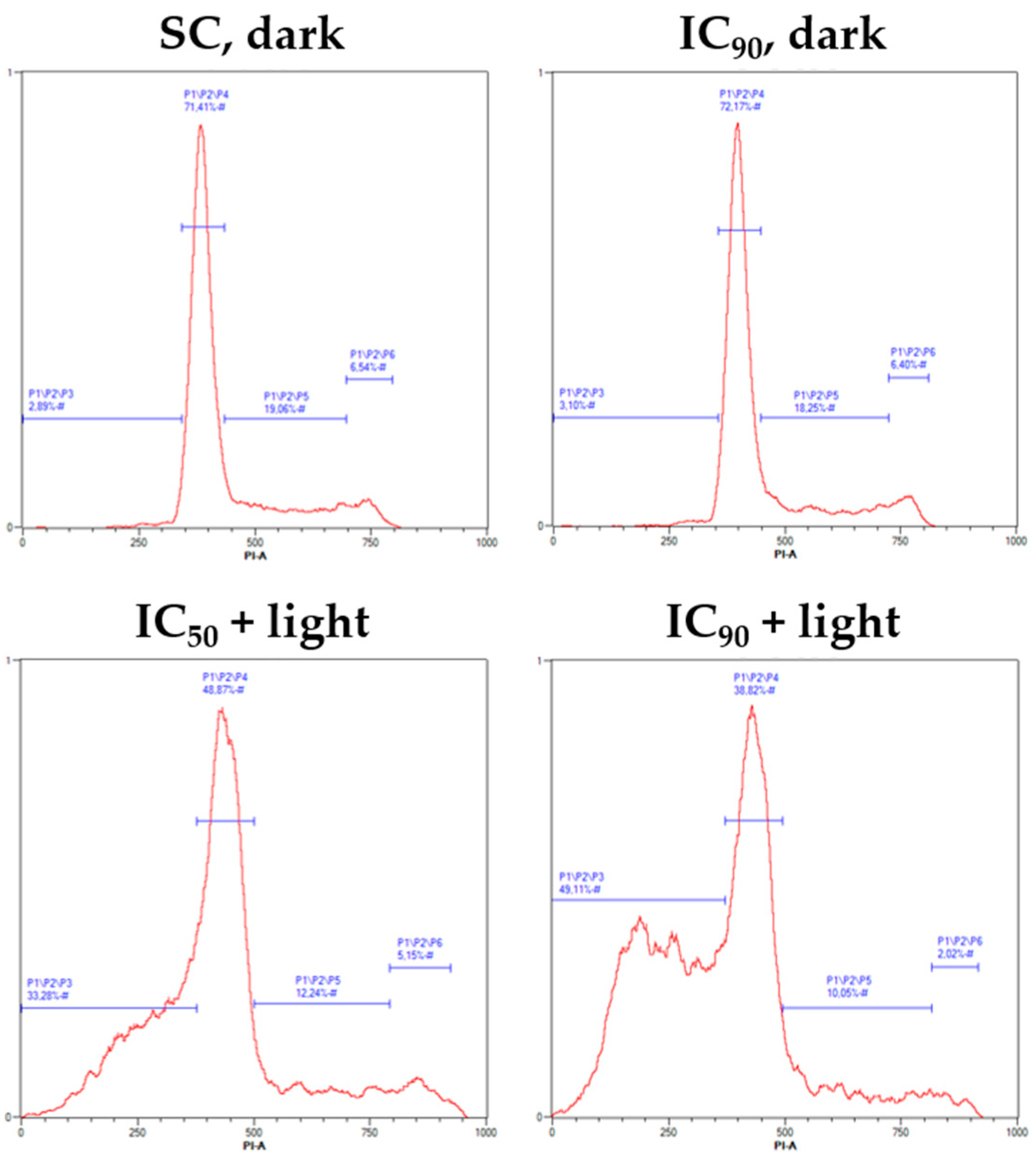
2.4. mTHPC-PDT Induces G2/M Arrest and the Formation of Sub G1 Populations with Fragmented DNA, Emphasizing the Induction of Apoptosis
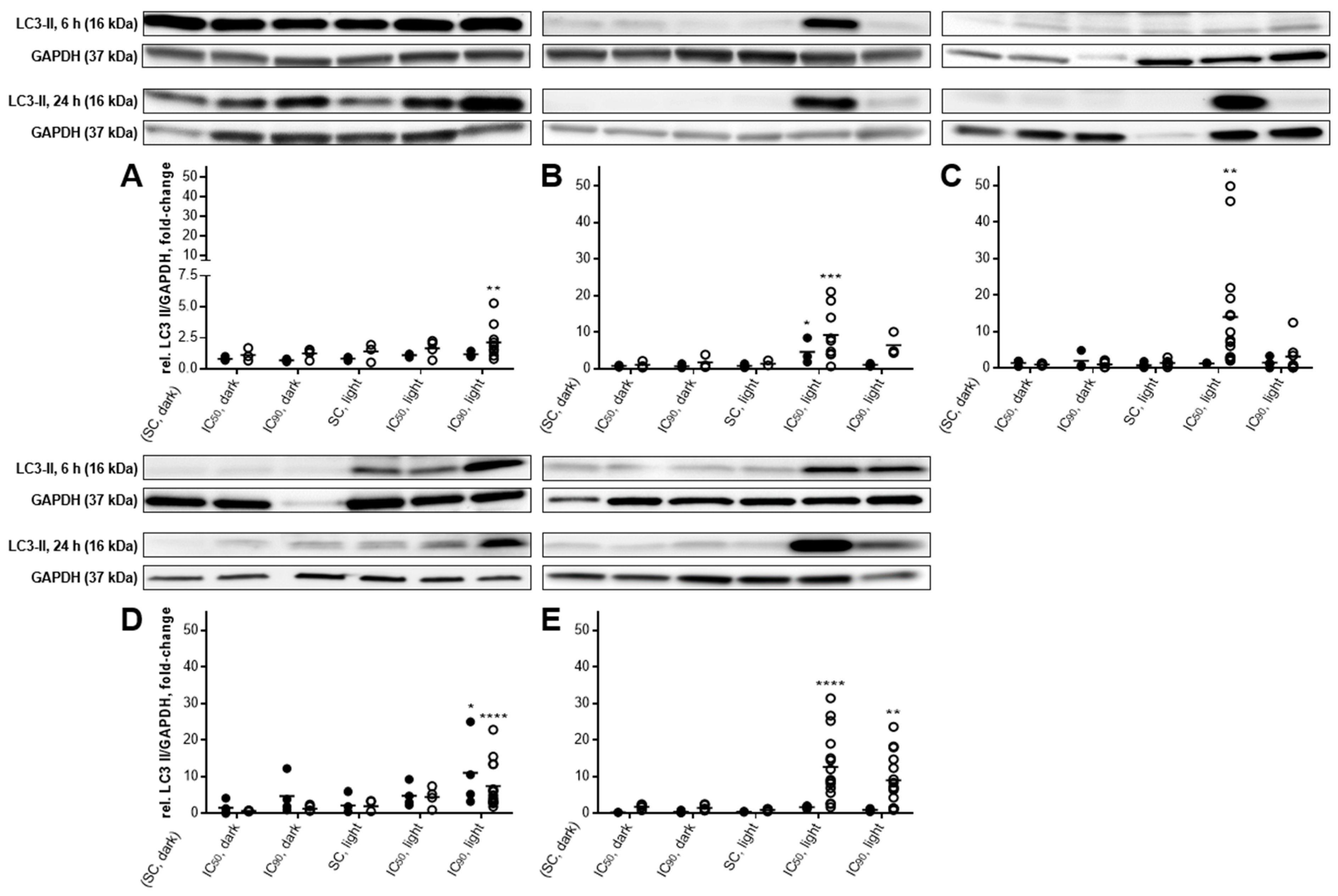
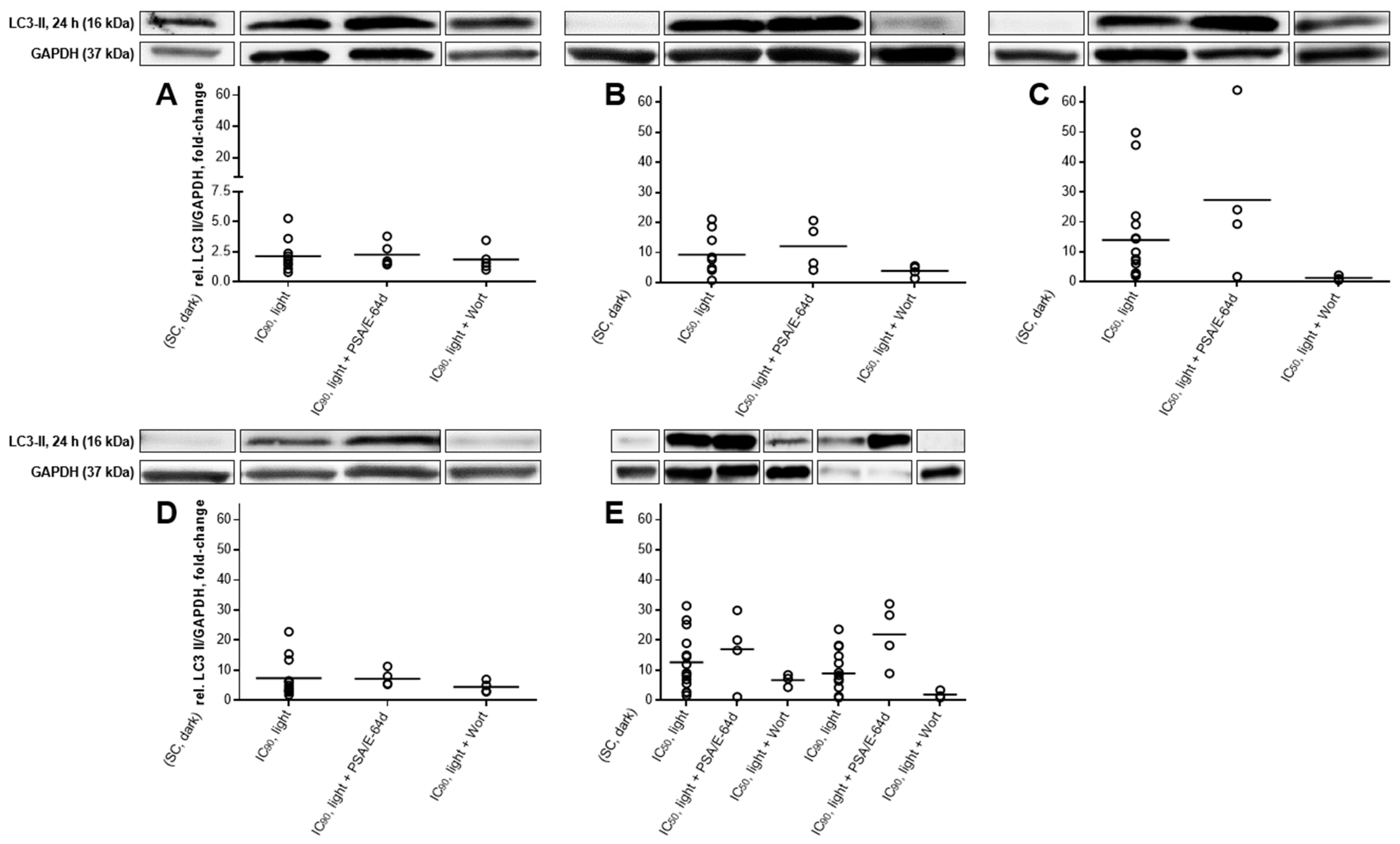
2.5. Autophagic Flux Analysis by LC3-II Levels in the Absence and Presence of Lysosomal Protease Inhibitors Revealed a Simultaneous Occurrence of Autophagy and Apoptosis after mTHPC-PDT
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Photosensitizer Treatment
4.3. Photodynamic Treatment
4.4. MTT Cell Viability Assay
4.5. LDH Release Assay
4.6. Flow Cytometry
4.6.1. Analysis of ROS Generation
4.6.2. Detection of Lipid Peroxidation (LPO)
4.6.3. Phosphatidylserine Externalization
4.6.4. Cell Cycle Analysis
4.7. Fluorescence Microscopy
4.8. Western Blotting
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCCP | Carbonyl Cyanide m-Chloro Phenyl Hydrazone |
| DCF | 2′,7′-Dichlorofluorescein |
| DOXO | Doxorubicin |
| FBS | Fetal Bovine Serum |
| GPX | Glutathione Peroxidase |
| GSH | Glutathione |
| GSR | Glutathione-disulfide Reductase |
| H2DCF-DA | 2′,7′-Dichlorodihydrofluorescein Diacetate |
| INT | 2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium Chloride |
| LDH | Lactate Dehydrogenase |
| LED | Light-emitting Diode |
| LPO | Lipid Peroxidation |
| mTHPC mTHPC-PDT | 5,10,15,20-tetra(m-hydroxyphenyl)chlorin mTHPC induced Photodynamic Therapy |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium Bromide |
| PARP | Poly(ADP-Ribose) Polymerase |
| PBS | Phosphate Buffered Saline |
| PDT | Photodynamic Therapy |
| PI | Propidium Iodide |
| PpIX | Protoporphyrin IX |
| PS | Photosensitizer |
| ROS | Reactive Oxygen Species |
| SC | Solvent-treated Control |
| t-BHP | tert-Butyl Hydroperoxide |
Appendix A






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triton X-100 | % Cellular Viability ± SD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dark | +Light | |||||||||
| A-427 | BHY | KYSE-70 | RT-4 | SISO | A-427 | BHY | KYSE-70 | RT-4 | SISO | |
| 0.1%, 6 h | 5.5 | 2.6 | 0 | 0.3 | 0.5 | 1.9 | 3.1 | 0.1 | 0.3 | 0.3 |
| ±9.0 | ±3.9 | + 0.7 | ±0.3 | ±0.5 | ±2.9 | ±5.0 | ±0.6 | ±0.4 | ±0.2 | |
| 0.01%, 24 h | 1.7 | 2.6 | 5.8 | 0.2 | 3.2 | 3.0 | 2.5 | 5.1 | 0.3 | 2.4 |
| ±2.7 | ±3.3 | ±5.2 | ±0.6 | ±2.4 | ±2.5 | ±3.3 | ±4.7 | ±0.4 | ±1.6 | |
References
- Lange, C.; Bednarski, P.J. Photosensitizers for photodynamic therapy: Photochemistry in the service of oncology. Curr. Pharm. Des. 2016, 22, 6956–6974. [Google Scholar] [CrossRef]
- Allison, R.R.; Sibata, C.H. Oncologic photodynamic therapy photosensitizers: A clinical review. Photodiagn. Photodyn. Ther. 2010, 7, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.B.; Brown, E.A.; Walker, I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004, 5, 497–508. [Google Scholar] [CrossRef]
- Bonnett, R.; White, R.D.; Winfield, U.J.; Berenbaum, M.C. Hydroporphyrins of the meso-tetra(hydroxyphenyl)porphyrin series as tumour photosensitizers. Biochem. J. 1989, 261, 277–280. [Google Scholar] [CrossRef] [Green Version]
- De Visscher, S.A.; Dijkstra, P.U.; Tan, I.B.; Roodenburg, J.L.; Witjes, M.J. mTHPC mediated photodynamic therapy (PDT) of squamous cell carcinoma in the head and neck: A systematic review. Oral Oncol. 2013, 49, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Senge, O.; Brandt, J.C. Temoporfin (Foscan®, 5,10,15,20-tetra(m-hydroxyphenyl)chlorin)—A second-generation photosensitizer. Photochem. Photobiol. 2011, 87, 1240–1296. [Google Scholar] [CrossRef]
- Marchal, S.; Dolivet, G.; Lassalle, H.-P.; Guillemin, F.; Bezdetnaya, L. Targeted photodynamic therapy in head and neck squamous cell carcinoma: Heading into the future. Lasers Med. Sci. 2015, 30, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, H.J.; Vernon, D.I.; Brown, S.B. Identification and partial characterization of an unusual distribution of the photosensitizer meta-tetrahydroxyphenyl chlorin (temoporfin) in human plasma. Photochem. Photobiol. 1999, 69, 482–488. [Google Scholar] [CrossRef]
- Civantos, F.J.; Karakullukcu, B.; Biel, M.; Silver, C.E.; Rinaldo, A.; Saba, N.F.; Ferlito, A. A review of photodynamic therapy for neoplasms of the head and neck. Adv. Ther. 2018, 35, 324–340. [Google Scholar] [CrossRef]
- Nelke, K.H.; Pawlak, W.; Leszczyszyn, J.; Gerber, H. Photodynamic therapy in head and neck cancer. Postepy Hig. Med. Dosw. 2014, 68, 119–128. [Google Scholar] [CrossRef]
- Reuther, T.; Kubler, A.C.; Zillmann, U.; Flechtenmacher, C.; Sinn, H. Comparison of the in vivo efficiency of Photofrin II-, mTHPC-, mTHPC-PEG- and mTHPCnPEG-mediated PDT in a human xenografted head and neck carcinoma. Lasers Surg. Med. 2001, 29, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.; Botter, S.M.; Campanile, C.; Robl, B.; Gräfe, S.; Pellegrini, G.; Fuchs, B. Foscan and foslip based photodynamic therapy in osteosarcoma in vitro and in intratibial mouse models. Int. J. Cancer 2017, 140, 1680–1692. [Google Scholar] [CrossRef]
- Gheewala, T.; Skwor, T.; Munirathinam, G. Photosensitizers in prostate cancer therapy. Oncotarget 2017, 8, 30524–30538. [Google Scholar] [CrossRef] [Green Version]
- Wachowska, M.; Osiak, A.; Muchowicz, A.; Gabrysiak, M.; Domagala, A.; Kilarski, W.W.; Golab, J. Investigation of cell death mechanisms in human lymphatic endothelial cells undergoing photodynamic therapy. Photodiagn. Photodyn. Ther. 2016, 14, 57–65. [Google Scholar] [CrossRef]
- François, A.; Marchal, S.; Guillemin, F.; Bezdetnaya, L. mTHPC-based photodynamic therapy induction of autophagy and apoptosis in cultured cells in relation to mitochondria and endoplasmic reticulum stress. Int. J. Oncol. 2011, 39, 1537–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchal, S.; François, A.; Dumas, D.; Guillemin, F.; Bezdetnaya, L. Relationship between subcellular localisation of Foscan and caspase activation in photosensitised MCF-7 cells. Br. J. Cancer 2007, 96, 944–951. [Google Scholar] [CrossRef]
- Kiesslich, T.; Berlanda, J.; Plaetzer, K.; Krammer, B.; Berr, F. Comparative characterization of the efficiency and cellular pharmacokinetics of Foscan- and Foslip-based photodynamic treatment in human biliary tract cancer cell lines. Photochem. Photobiol. Sci. 2007, 6, 619–627. [Google Scholar] [CrossRef]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef]
- Kessel, D. PDT: Death and survival pathways. In Handbook of Photodynamic Therapy: Updates on Recent Applications of Porphyrin-Based Compounds; Pandey, R.K., Kessel, D., Dougherty, T.J., Eds.; World Scientific: Hackensack, NJ, USA, 2016; pp. 319–333. [Google Scholar] [CrossRef]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2’,7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef]
- Raudsepp, P.; Brüggemann, D.A.; Andersen, M.L. Detection of radicals in single droplets of oil-in-water emulsions with the lipophilic fluorescent probe BODIPY665/676 and confocal laser scanning microscopy. Free Radic. Biol. Med. 2014, 70, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Drummen, G.P.C.; van Liebergen, L.C.M.; op den Kamp, J.A.F.; Post, J.A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Richter, C. Mitochondria as targets for the induction of apoptosis in photodynamic therapy. In Photomedicine in Gynecology and Reproduction; Wyss, P., Tadir, Y., Tromberg, B.J., Haller, U., Eds.; S. Karger: Basel, Switzerland, 2000; pp. 157–168. [Google Scholar] [CrossRef]
- Satoh, T.; Enokido, Y.; Aoshima, H.; Uchiyama, Y.; Hatanaka, H. Changes in mitochondrial membrane potential during oxidative stress-induced apoptosis in PC12 cells. J. Neurosci. Res. 1997, 50, 413–420. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two—Cellular signaling, cell metabolism and modes of cell death. Photodiagn. Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. 2002, 1, 1–21. [Google Scholar] [CrossRef]
- Yu, S.-W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Nagase, H.; Kawane, K.; Mukae, N.; Fukuyama, H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003, 10, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Zughaier, S.M. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelarova, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Berlanda, J.; Kiesslich, T.; Engelhardt, V.; Krammer, B.; Plaetzer, K. Comparative in vitro study on the characteristics of different photosensitizers employed in PDT. J. Photochem. Photobiol. B 2010, 100, 173–180. [Google Scholar] [CrossRef]
- Abdulrehman, G.; Xv, K.; Li, Y.; Kang, L. Effects of meta-tetrahydroxyphenylchlorin photodynamic therapy on isogenic colorectal cancer SW480 and SW620 cells with different metastatic potentials. Lasers Med. Sci. 2018, 33, 1581–1590. [Google Scholar] [CrossRef]
- Löw, K.; Knobloch, T.; Wagner, S.J.; Wiehe, A.; Engel, A.; Langer, K.; von Briesen, H. Comparison of intracellular accumulation and cytotoxicity of freemTHPC andmTHPC-loaded PLGA nanoparticles in human colon carcinoma cells. Nanotechnology 2011, 22, 245102. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Golab, J. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Girotti, A.W. Photosensitized oxidation of membrane lipids: Reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J. Photochem. Photobiol. B 2001, 63, 103–113. [Google Scholar] [CrossRef]
- Bilski, P.; Belanger, A.G.; Chignell, C.F. Photosensitized oxidation of 2’,7’-dichlorofluorescin: Singlet oxygen does not contribute to the formation of fluorescent oxidation product 2’,7’-dichlorofluorescein. Free Radic. Biol. Med. 2002, 33, 938–946. [Google Scholar] [CrossRef]
- Redmond, R.W.; Gamlin, J.N. A compilation of singlet oxygen yields from biologically relevant molecules. Photochem. Photobiol. 1999, 70, 391–475. [Google Scholar] [CrossRef]
- Bonnett, R.; Charlesworth, P.; Djelal, B.D.; Foley, S.; McGarvey, D.J.; Truscott, T.G. Photophysical properties of 5,10,15,20-tetrakis(m-hydroxyphenyl)porphyrin (m-THPP), 5,10,15,20-tetrakis(m-hydroxyphenyl)chlorin (m-THPC) and 5,10,15,20-tetrakis(m-hydroxyphenyl)bacteriochlorin (m-THPBC): A comparative study. J. Chem. Soc. Perkin Trans. 2 1999, 2, 325–328. [Google Scholar] [CrossRef]
- Bracht, K.; Boubakari; Grünert, R.; Bednarski, P.J. Correlations between the activities of 19 anti-tumor agents and the intracellular glutathione concentrations in a panel of 14 human cancer cell lines: Comparisons with the National Cancer Institute data. Anticancer Drugs 2006, 17, 41–51. [Google Scholar] [CrossRef]
- Ehrenberg, B.; Gross, E.; Nitzan, Y.; Malik, Z. Electric depolarization of photosensitized cells: Lipid vs. protein alterations. Biochim. Biophys. Acta 1993, 1151, 257–264. [Google Scholar] [CrossRef]
- Gaullier, J.M.; Valla, A.; Bazin, M.; Giraud, M.; Dubertret, L.; Santus, R. N-conjugates of 2,5-disubstituted pyrrole and glutathione. Evaluation of their potency as antioxidants against photosensitization of NCTC 2544 keratinocytes by excess endogenous protoporphyrin IX. J. Photochem. Photobiol. B 1997, 39, 24–29. [Google Scholar] [CrossRef]
- Klein, S.D.; Walt, H.; Richter, C. Photosensitization of isolated rat liver mitochondria by tetra(m-hydroxyphenyl)chlorin. Arch. Biochem. Biophys. 1997, 348, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.O.; Bezdetnaya, L.N.; Potapenko, A.Y.; Guillemin, F. Photodynamic properties of meta-tetra(hydroxyphenyl)chlorin in human tumor cells. Radiat. Res. 1999, 152, 428–435. [Google Scholar] [CrossRef]
- Kirveliene, V.; Prasmickaite, L.; Kadziauskas, J.; Bonnett, R.; Djelal, B.D.; Juodka, B. Post-exposure processes in Temoporfin-photosensitized cells in vitro: Reliance on energy metabolism. J. Photochem. Photobiol. B 1997, 41, 173–180. [Google Scholar] [CrossRef]
- Marchal, S.; Fadloun, A.; Maugain, E.; D’Hallewin, M.A.; Guillemin, F.; Bezdetnaya, L. Necrotic and apoptotic features of cell death in response to Foscan photosensitization of HT29 monolayer and multicell spheroids. Biochem. Pharmacol. 2005, 69, 1167–1176. [Google Scholar] [CrossRef]
- Marchal, S.; Bezdetnaya, L.; Guillemin, F. Modality of cell death induced by Foscan-based photodynamic treatment in human colon adenocarcinoma cell line HT29. Biochemistry (Mosc.) 2004, 69, 45–49. [Google Scholar] [CrossRef]
- Yow, C.M.N.; Mak, N.K.; Leung, A.W.N.; Huang, Z. Induction of early apoptosis in human nasopharyngeal carcinoma cells by mTHPC-mediated photocytotoxicity. Photodiagn. Photodyn. Ther. 2009, 6, 122–127. [Google Scholar] [CrossRef]
- Segawa, K.; Nagata, S. An apoptotic ‘me’ signal: Phosphatidylserine exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [Green Version]
- Mandal, D.; Moitra, P.K.; Saha, S.; Basu, J. Caspase 3 regulates phosphatidylserine externalization and phagocytosis of oxidatively stressed erythrocytes. FEBS Lett. 2002, 513, 184–188. [Google Scholar] [CrossRef]
- Boucher, D.; Blais, V.; Denault, J.-B. Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc. Natl. Acad. Sci. USA 2012, 109, 5669–5674. [Google Scholar] [CrossRef] [Green Version]
- D’Amours, D.; Sallmann, F.R.; Dixit, V.M.; Poirier, G.G. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: Implications for apoptosis. J. Cell Sci. 2001, 114, 3771–3778. [Google Scholar]
- Puig, B.; Tortosa, A.; Ferrer, I. Cleaved caspase-3, caspase-7 and poly (ADP-ribose) polymerase are complementarily but differentially expressed in human medulloblastomas. Neurosci. Lett. 2001, 306, 85–88. [Google Scholar] [CrossRef]
- Wyrsch, P.; Blenn, C.; Bader, J.; Althaus, F.R. Cell death and autophagy under oxidative stress: Roles of poly(ADP-ribose) polymerases and Ca2+. Mol. Cell. Biol. 2012, 32, 3541–3553. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, Y.; Jiang, R.; Li, E.; Chen, X.; Xi, Z.; Jiang, X. PARP and RIP 1 are required for autophagy induced by 11’-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 2011, 7, 598–612. [Google Scholar] [CrossRef]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef]
- Herceg, Z.; Wang, Z.-Q. Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Mol. Cell. Biol. 1999, 19, 5124–5133. [Google Scholar] [CrossRef]
- Bacellar, I.O.; Tsubone, T.M.; Pavani, C.; Baptista, M.S. Photodynamic efficiency: From molecular photochemistry to cell death. Int. J. Mol. Sci. 2015, 16, 20523–20559. [Google Scholar] [CrossRef]
- Mroz, P.; Yaroslavsky, A.; Kharkwal, G.B.; Hamblin, M.R. Cell death pathways in photodynamic therapy of cancer. Cancers 2011, 3, 2516–2539. [Google Scholar] [CrossRef]
- Gharehbaghi, K.; Kubin, A.; Grusch, M.; Gharehbaghi-Schnell, E.; Wierrani, F.; Jayaram, H.N.; Szekeres, T. Photodynamic action of meta-tetrahydroxyphenylchlorin (mTHPC) on an ovarian cancer cell line. Anticancer Res. 2000, 20, 2647–2652. [Google Scholar] [PubMed]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. In Methods in Cell Biology: Recent Advances in Cytometry, Part B; Darzynkiewicz, Z., Holden, E., Orfao, A., Telford, W., Wlodkowic, D., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 103, pp. 55–98. [Google Scholar] [CrossRef]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phosphorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Claybon, A.; Karia, B.; Bruce, C.; Bishop, A.J.R. PARP1 suppresses homologous recombination events in mice in vivo. Nucleic Acids Res. 2010, 38, 7538–7545. [Google Scholar] [CrossRef] [Green Version]
- Kukcinaviciute, E.; Sasnauskiene, A.; Dabkeviciene, D.; Kirveliene, V.; Jonusiene, V. Effect of mTHPC-mediated photodynamic therapy on 5-fluorouracil resistant human colorectal cancer cells. Photochem. Photobiol. Sci. 2017, 16, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Bezdetnaya, L.; Morliere, P.; Santus, R.; Guillemin, F. Endoplasmic reticulum and Golgi apparatus are the preferential sites of Foscan® localisation in cultured tumour cells. Br. J. Cancer 2003, 88, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Mak, N.K.; Yow, C.M.; Fung, M.C.; Chiu, L.C.; Leung, W.N.; Cheung, N.H. The binding characteristics and intracellular localization of temoporfin (mTHPC) in myeloid leukemia cells: Phototoxicity and mitochondrial damage. Photochem. Photobiol. 2000, 72, 541–547. [Google Scholar] [CrossRef]
- Inguscio, V.; Panzarini, E.; Dini, L. Autophagy contributes to the death/survival balance in cancer photodynamic therapy. Cells 2012, 1, 464–491. [Google Scholar] [CrossRef]
- Andrzejak, M.; Price, M.; Kessel, D.H. Apoptotic and autophagic responses to photodynamic therapy in 1c1c7 murine hepatoma cells. Autophagy 2011, 7, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Reiners, J.J.; Agostinis, P.; Berg, K.; Oleinick, N.L.; Kessel, D. Assessing autophagy in the context of photodynamic therapy. Autophagy 2010, 6, 7–18. [Google Scholar] [CrossRef]
- Sasnauskiene, A.; Kadziauskas, J.; Vezelyte, N.; Jonusiene, V.; Kirveliene, V. Apoptosis, autophagy and cell cycle arrest following photodamage to mitochondrial interior. Apoptosis 2009, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Pieslinger, A.; Plaetzer, K.; Oberdanner, C.B.; Berlanda, J.; Mair, H.; Krammer, B.; Kiesslich, T. Characterization of a simple and homogeneous irradiation device based on light-emitting diodes: A possible low-cost supplement to conventional light sources for photodynamic treatment. Med. Laser Appl. 2006, 21, 277–283. [Google Scholar] [CrossRef]
- Lange, C.; Bednarski, P.J. Evaluation for synergistic effects by combinations of photodynamic therapy (PDT) with temoporfin (mTHPC) and Pt(II) complexes carboplatin, cisplatin or oxaliplatin in a set of five human cancer cell lines. Int. J. Mol. Sci. 2018, 19, 3183. [Google Scholar] [CrossRef]
- Haidara, K.; Morel, I.; Abalea, V.; Gascon Barre, M.; Denizeau, F. Mechanism of tert-butylhydroperoxide induced apoptosis in rat hepatocytes: Involvement of mitochondria and endoplasmic reticulum. Biochim. Biophys. Acta 2002, 1542, 173–185. [Google Scholar] [CrossRef]
- Rebbaa, A.; Zheng, X.; Chou, P.M.; Mirkin, B.L. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene 2003, 22, 2805–2811. [Google Scholar] [CrossRef] [Green Version]
- Skladanowski, A.; Konopa, J. Adriamycin and daunomycin induce programmed cell death (apoptosis) in tumour cells. Biochem. Pharmacol. 1993, 46, 375–382. [Google Scholar] [CrossRef]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Shen, X.; Xiao, X.L.; Liu, M.Y.; Li, S.L.; Yan, J.; Dong, D.L. Mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone induces vasorelaxation without involving KATP channel activation in smooth muscle cells of arteries. Br. J. Pharmacol. 2016, 173, 3145–3158. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]











| Dark | 1.8 J/cm2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A-427 | BHY | KYSE-70 | RT-4 | SISO | A-427 | BHY | KYSE-70 | RT-4 | SISO | |
| IC50 (µM) | 0.6 | 1.0 | 1.0 | 1.8 | 1.0 | 0.02 | 0.06 | 0.06 | 0.1 | 0.06 |
| IC90 (µM) | 1.4 | 2.8 | 2.8 | 3.2 | 1.9 | 0.07 | 0.1 | 0.2 | 0.3 | 0.1 |
| Triton X-100 | LDH Release (% Cytotoxicity ± SD) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dark | +Light | |||||||||
| A-427 | BHY | KYSE-70 | RT-4 | SISO | A-427 | BHY | KYSE-70 | RT-4 | SISO | |
| 0.1%, 6 h | 107.4 | 97.1 | 81.7 | 104.2 | 98.1 | 111.2 | 95.3 | 99.5 | 102.0 | 101.8 |
| ±14.8 | ±3.8 | ±30.6 | ±10.2 | ±10.0 | ±23.4 | ±7.9 | ±18.7 | ±7.8 | ±10.8 | |
| 0.01%, 24 h | 59.7 | 57.9 | 66.5 | 57.7 | 70.3 | 45.2 | 51.1 | 63.2 | 47.5 | 75.3 |
| ±13.3 | ±6.7 | ±12.7 | ±10.1 | ±14.9 | ±11.6 | ±2.8 | ±17.4 | ±5.4 | ±29.6 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lange, C.; Lehmann, C.; Mahler, M.; Bednarski, P.J. Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines. Cancers 2019, 11, 702. https://doi.org/10.3390/cancers11050702
Lange C, Lehmann C, Mahler M, Bednarski PJ. Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines. Cancers. 2019; 11(5):702. https://doi.org/10.3390/cancers11050702
Chicago/Turabian StyleLange, Carsten, Christiane Lehmann, Martin Mahler, and Patrick J. Bednarski. 2019. "Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines" Cancers 11, no. 5: 702. https://doi.org/10.3390/cancers11050702
APA StyleLange, C., Lehmann, C., Mahler, M., & Bednarski, P. J. (2019). Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines. Cancers, 11(5), 702. https://doi.org/10.3390/cancers11050702