Platelets as Key Factors in Hepatocellular Carcinoma


{kind=link}
{kind=link}
Abstract
:1. Introduction
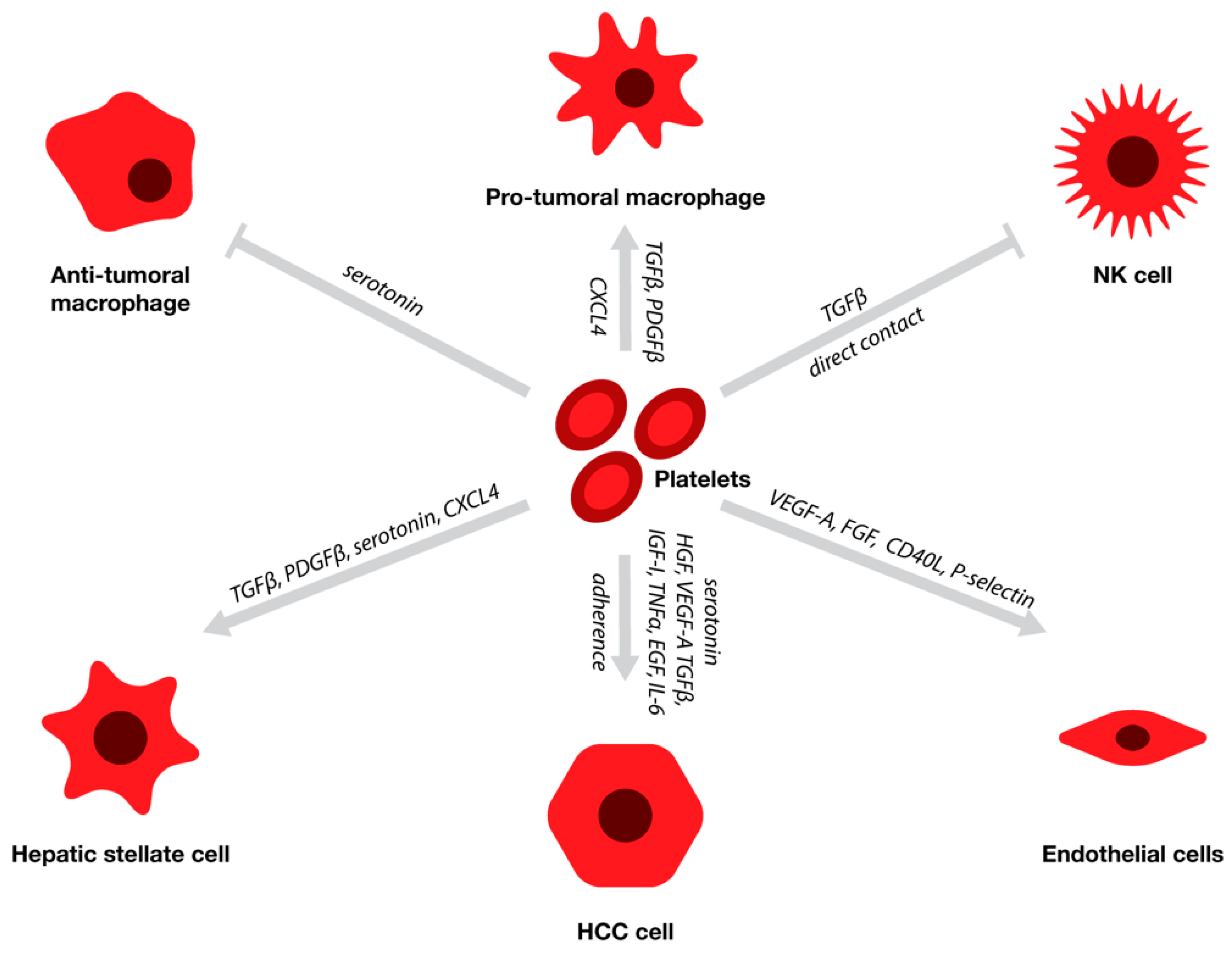
2. The Effect of Platelets on HCC Proliferation and Metastasis
3. Platelets, Liver Sinusoidal Endothelial Cells, and Angiogenesis
4. The Role of Platelets in HSC Activation
5. Platelets and the Hepatic Immune Response
6. Thrombocytosis and Thrombocytopenia in Liver Disease
7. Targeting Platelets as a Therapeutic Strategy in Liver Disease
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The Role of Inflammation in Liver Cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar] [CrossRef] [PubMed]
- Coulon, S.; Heindryckx, F.; Geerts, A.; Van Steenkiste, C.; Colle, I.; Van Vlierberghe, H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2011, 31, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Van de Veire, S.; Stalmans, I.; Heindryckx, F.; Oura, H.; Tijeras-Raballand, A.; Schmidt, T.; Loges, S.; Albrecht, I.; Jonckx, B.; Vinckier, S.; et al. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell 2010, 141, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Best, J.; Verhulst, S.; Syn, W.K.; Lagaisse, K.; van Hul, N.; Heindryckx, F.; Sowa, J.P.; Peeters, L.; Van Vlierberghe, H.; Leclercq, I.A.; et al. Macrophage Depletion Attenuates Extracellular Matrix Deposition and Ductular Reaction in a Mouse Model of Chronic Cholangiopathies. PLoS ONE 2016, 11, e0162286. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaff, D.; Kwanten, W.J.; Couturier, F.J.; Govaerts, J.S.; Verlinden, W.; Brosius, I.; D’Hondt, M.; Driessen, A.; De Winter, B.Y.; De Man, J.G.; et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab. Investig. 2018, 98, 1263–1275. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Vignoli, A.; Balducci, D. Clotting mechanisms and cancer: Implications in thrombus formation and tumor progression. Clin. Adv. Hematol. Oncol. 2003, 1, 673–678. [Google Scholar]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed. Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Menter, D.G.; Tucker, S.C.; Kopetz, S.; Sood, A.K.; Crissman, J.D.; Honn, K.V. Platelets and cancer: A casual or causal relationship: Revisited. Cancer Metast. Rev. 2014, 33, 231–269. [Google Scholar] [CrossRef]
- Zanetto, A.; Campello, E.; Spiezia, L.; Burra, P.; Simioni, P.; Russo, F.P. Cancer-Associated Thrombosis in Cirrhotic Patients with Hepatocellular Carcinoma. Cancers 2018, 10, 450. [Google Scholar] [CrossRef]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.W.; Chung, J.; Sverdlov, D.Y.; Miyamoto, M.; Kim, Y.O.; Ogawa, S.; Arch, R.H.; et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014, 147, 1378–1392. [Google Scholar] [CrossRef]
- Maini, M.K.; Schurich, A. Platelets harness the immune response to drive liver cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 12840–12841. [Google Scholar] [CrossRef] [Green Version]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef]
- Sitia, G.; Iannacone, M.; Guidotti, L.G. Anti-platelet therapy in the prevention of hepatitis B virus-associated hepatocellular carcinoma. J. Hepatol. 2013, 59, 1135–1138. [Google Scholar] [CrossRef] [Green Version]
- Carr, B.I.; Cavallini, A.; D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Mazzocca, A.; Messa, C. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer 2014, 14, 43. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers 2019, 11, 24. [Google Scholar] [CrossRef]
- Myronovych, A.; Murata, S.; Chiba, M.; Matsuo, R.; Ikeda, O.; Watanabe, M.; Hisakura, K.; Nakano, Y.; Kohno, K.; Kawasaki, T.; et al. Role of platelets on liver regeneration after 90% hepatectomy in mice. J. Hepatol. 2008, 49, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, R.; Ohkohchi, N.; Murata, S.; Ikeda, O.; Nakano, Y.; Watanabe, M.; Hisakura, K.; Myronovych, A.; Kubota, T.; Narimatsu, H.; et al. Platelets Strongly Induce Hepatocyte Proliferation with IGF-1 and HGF In Vitro. J. Surg. Res. 2008, 145, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, R.; Nakano, Y.; Ohkohchi, N. Platelet Administration Via the Portal Vein Promotes Liver Regeneration in Rats After 70% Hepatectomy. Ann. Surg. 2011, 253, 759–763. [Google Scholar] [CrossRef] [Green Version]
- He, A.D.; Xie, W.; Song, W.; Ma, Y.Y.; Liu, G.; Liang, M.L.; Da, X.W.; Yao, G.Q.; Zhang, B.X.; Gao, C.J.; et al. Platelet releasates promote the proliferation of hepatocellular carcinoma cells by suppressing the expression of KLF6. Sci. Rep. 2017, 7, 3989. [Google Scholar] [CrossRef]
- Hoshi, R.; Murata, S.; Matsuo, R.; Myronovych, A.; Hashimoto, I.; Ikeda, H.; Ohkohchi, N. Freeze-dried platelets promote hepatocyte proliferation in mice. Cryobiology 2007, 55, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Ohkohchi, N.; Matsuo, R.; Ikeda, O.; Myronovych, A.; Hoshi, R. Platelets promote liver regeneration in early period after hepatectomy in mice. World J. Surg. 2007, 31, 808–816. [Google Scholar] [CrossRef]
- Lalor, P.F.; Herbert, J.; Bicknell, R.; Adams, D.H. Hepatic sinusoidal endothelium avidly binds platelets in an integrin-dependent manner, leading to platelet and endothelial activation and leukocyte recruitment. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G469–G478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Murata, S.; Takahashi, K.; Nozaki, R.; Ohshiro, Y.; Ikeda, N.; Pak, S.; Myronovych, A.; Hisakura, K.; Fukunaga, K.; et al. Activation of human liver sinusoidal endothelial cell by human platelets induces hepatocyte proliferation. J. Hepatol. 2010, 53, 648–654. [Google Scholar] [CrossRef] [Green Version]
- Selzner, N.; Selzner, M.; Odermatt, B.; Tian, Y.H.; Van Rooijen, N.; Clavien, P.A. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-alpha/IL-6 in mice. Gastroenterology 2003, 124, 692–700. [Google Scholar] [CrossRef]
- Takahashi, K.; Kozuma, Y.; Suzuki, H.; Tamura, T.; Maruyama, T.; Fukunaga, K.; Murata, S.; Ohkohchi, N. Human platelets promote liver regeneration with Kupffer cells in SCID mice. J. Surg. Res. 2013, 180, 62–72. [Google Scholar] [CrossRef]
- Sharma, D.; Brummel-Ziedins, K.E.; Bouchard, B.A.; Holmes, C.E. Platelets in Tumor Progression: A Host Factor That Offers Multiple Potential Targets in the Treatment of Cancer. J. Cell. Physiol. 2014, 229, 1005–1015. [Google Scholar] [CrossRef]
- Marx, S.; Xiao, Y.; Baschin, M.; Splittstohser, M.; Altmann, R.; Moritz, E.; Jedlitschky, G.; Bien-Moller, S.; Schroeder, H.W.S.; Rauch, B.H. The Role of Platelets in Cancer Pathophysiology: Focus on Malignant Glioma. Cancers 2019, 11, 569. [Google Scholar] [CrossRef]
- Morimoto, Y.; Nouso, K.; Wada, N.; Takeuchi, Y.; Kinugasa, H.; Miyahara, K.; Yasunaka, T.; Kuwaki, K.; Onishi, H.; Ikeda, F.; et al. Involvement of platelets in extrahepatic metastasis of hepatocellular carcinoma. Hepatol. Res. 2014, 44, E353–E359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavergne, M.; Janus-Bell, E.; Schaff, M.; Gachet, C.; Mangin, P.H. Platelet Integrins in Tumor Metastasis: Do They Represent a Therapeutic Target? Cancers 2017, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Coupland, L.A.; Chong, B.H.; Parish, C.R. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res. 2012, 72, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Xin, G.; Wei, Z.; Li, S.; Xing, Z.; Ji, C.; Du, J.; Niu, H.; Huang, W. Dihydrodiosgenin inhibits endothelial cell-derived factor VIII and platelet-mediated hepatocellular carcinoma metastasis. Cancer Manag. Res. 2019, 11, 4871–4882. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Giannuzzi, G.; Carella, N.; Messa, C.; Cavallini, A.; Carr, B.I. Antagonism of sorafenib and regorafenib actions by platelet factors in hepatocellular carcinoma cell lines. BMC Cancer 2014, 14, 351. [Google Scholar] [CrossRef]
- Radziwon-Balicka, A.; Medina, C.; O’Driscoll, L.; Treumann, A.; Bazou, D.; Inkielewicz-Stepniak, I.; Radomski, A.; Jow, H.; Radomski, M.W. Platelets increase survival of adenocarcinoma cells challenged with anticancer drugs: Mechanisms and implications for chemoresistance. Br. J. Pharmacol. 2012, 167, 787–804. [Google Scholar] [CrossRef]
- Ishikawa, S.; Miyashita, T.; Inokuchi, M.; Hayashi, H.; Oyama, K.; Tajima, H.; Takamura, H.; Ninomiya, I.; Ahmed, A.K.; Harman, J.W.; et al. Platelets surrounding primary tumor cells are related to chemoresistance. Oncol. Rep. 2016, 36, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Herr, N.; Bode, C.; Duerschmied, D. The Effects of Serotonin in Immune Cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef]
- Liang, C.; Chen, W.; Zhi, X.; Ma, T.; Xia, X.F.; Liu, H.; Zhang, Q.; Hu, Q.D.; Zhang, Y.; Bai, X.L.; et al. Serotonin promotes the proliferation of serum-deprived hepatocellular carcinoma cells via upregulation of FOXO3a. Mol. Cancer 2013, 12, 14. [Google Scholar] [CrossRef]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin Promotes Tumor Growth in Human Hepatocellular Cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef]
- Shu, B.; Wang, S.; Deng, Y.; Zhai, M.M.; Liu, S.S. Intra-platelet serotonin in prognosis of tumorigenesis: Friend or foe? J. Hepatol. 2018, 68, 1333–1334. [Google Scholar] [CrossRef]
- Kong, L.M.; Yao, L.; Lu, N.; Dong, Y.L.; Zhang, J.; Wang, Y.Q.; Liu, L.; Zhang, H.L.; Huang, J.G.; Liao, C.G. Interaction of KLF6 and Sp1 regulates basigin-2 expression mediated proliferation, invasion and metastasis in hepatocellular carcinoma. Oncotarget 2016, 7, 27975–27987. [Google Scholar] [CrossRef]
- Heindryckx, F.; Gerwins, P. Targeting the tumor stroma in hepatocellular carcinoma. World J. Hepatol. 2015, 7, 165–176. [Google Scholar] [CrossRef]
- Choi, S.B.; Han, H.J.; Kim, W.B.; Song, T.J.; Choi, S.Y. VEGF Overexpression Predicts Poor Survival in Hepatocellular Carcinoma. Open Med. (Wars) 2017, 12, 430–439. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, J.; Luo, F. Serum tumor markers for detection of hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 1175–1181. [Google Scholar] [CrossRef]
- Poon, R.T.P.; Ng, I.O.L.; Lau, C.; Yu, W.C.; Fan, S.T.; Wong, J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am. J. Surg. 2001, 182, 298–304. [Google Scholar] [CrossRef]
- Schoenleber, S.J.; Kurtz, D.M.; Talwalker, J.A.; Roberts, L.R.; Gores, G.J. Prognostic role of vascular endothelial growth factor in hepatocellular carcinoma: Systematic review and meta-analysis. Brit. J. Cancer 2009, 100, 1385–1392. [Google Scholar] [CrossRef]
- Ferroni, P.; Spila, A.; D’Alessandro, R.; Martini, F.; Iacovone, F.; Ettorre, G.M.; Vennarecci, G.; Santoro, R.; Puoti, C.; Guadagni, F. Platelet activation and vascular endothelial growth factor 165 release in hepatocellular cancer. Clin. Chim. Acta 2011, 412, 450–454. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metast. Rev. 2017, 36, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Semenov, A.V.; Romanov, Y.A.; Loktionova, S.A.; Tikhomirov, O.Y.; Khachikian, M.V.; Vasil’ev, S.A.; Mazurov, A.V. Production of soluble P-selectin by platelets and endothelial cells. Biochemistry (Mosc) 1999, 64, 1326–1335. [Google Scholar]
- Kohler, S.; Ullrich, S.; Richter, U.; Schumacher, U. E-/P-selectins and colon carcinoma metastasis: First in vivo evidence for their crucial role in a clinically relevant model of spontaneous metastasis formation in the lung. Br. J. Cancer 2010, 102, 602–609. [Google Scholar] [CrossRef]
- Ripoche, J. Blood platelets and inflammation: Their relationship with liver and digestive diseases. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 353–357. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.N.; Lim, S.Y.; Yuzhalin, A.E.; Jones, K.; Markelc, B.; Kim, K.J.; Buzzelli, J.N.; Fokas, E.; Cao, Y.; Smart, S.; et al. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology 2017, 65, 1920–1935. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Muller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- Croner, R.S.; Hoerer, E.; Kulu, Y.; Hackert, T.; Gebhard, M.M.; Herfarth, C.; Klar, E. Hepatic platelet and leukocyte adherence during endotoxemia. Crit. Care 2006, 10, R15. [Google Scholar] [CrossRef]
- Chakraborty, S.; Hong, W.J. Linking Extracellular Matrix Agrin to the Hippo Pathway in Liver Cancer and Beyond. Cancers 2018, 10, 45. [Google Scholar] [CrossRef]
- Pinzani, M. PDGF and signal transduction in hepatic stellate cells. Front. Biosci. 2002, 7, d1720–d1726. [Google Scholar] [CrossRef]
- Song, Y.; Kim, S.H.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci. Rep. 2016, 6, 36750. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.J.; Jain, G.; Rajagopalan, P. Designing a fibrotic microenvironment to investigate changes in human liver sinusoidal endothelial cell function. Acta Biomater. 2015, 24, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; van Roeyen, C.R.C.; Ostendorf, T.; Floege, J.; Gressner, A.M.; Weiskirchen, R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 2007, 46, 1064–1074. [Google Scholar] [CrossRef]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; Odell, M.M.; Bauer, R.L.; Ren, H.P.; Haugen, H.S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Yan, L.; Zhao, H.; Wang, G.; China Hep, B.R.F.A.R.G. Serum platelet-derived growth factor BB levels: A potential biomarker for the assessment of liver fibrosis in patients with chronic hepatitis B. Int. J. Infect. Dis. 2016, 49, 94–99. [Google Scholar] [CrossRef]
- Ruddell, R.G.; Oakley, F.; Hussain, Z.; Yeung, I.; Bryan-Lluka, L.J.; Ramm, G.A.; Mann, D.A. A role for serotonin (5-HT) in hepatic stellate cell function and liver fibrosis. Am. J. Pathol. 2006, 169, 861–876. [Google Scholar] [CrossRef]
- Maass, T.; Thieringer, F.R.; Mann, A.; Longerich, T.; Schirmacher, P.; Strand, D.; Hansen, T.; Galle, P.R.; Teufel, A.; Kanzler, S. Liver specific overexpression of platelet-derived growth factor-B accelerates liver cancer development in chemically induced liver carcinogenesis. Int. J. Cancer 2011, 128, 1259–1268. [Google Scholar] [CrossRef]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-beta1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef]
- Mahmoud, N.I.; Messiha, B.A.S.; Salehc, I.G.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Interruption of platelets and thrombin function as a new approach against liver fibrosis induced experimentally in rats. Life Sci. 2019. [Google Scholar] [CrossRef]
- Yabanoglu, S.; Akkiki, M.; Seguelas, M.H.; Mialet-Perez, J.; Parini, A.; Pizzinat, N. Platelet derived serotonin drives the activation of rat cardiac fibroblasts by 5-HT2A receptors. J. Mol. Cell. Cardiol. 2009, 46, 518–525. [Google Scholar] [CrossRef]
- Chen, L.P.; Chen, G.; Guo, Y.B.; Liu, L.; Xiao, L.; Fan, W.M.; Shi, B.Y.; Qian, Y.Y. Ketanserin, a Serotonin 2A Receptor Antagonist, Alleviates Ischemia-Related Biliary Fibrosis Following Donation After Cardiac Death Liver Transplantation in Rats. Liver Transplant. 2014, 20, 1317–1326. [Google Scholar] [CrossRef]
- Zaldivar, M.M.; Pauels, K.; von Hundelshausen, P.; Berres, M.L.; Schmitz, P.; Bornemann, J.; Kowalska, M.A.; Gassler, N.; Streetz, K.L.; Weiskirchen, R.; et al. CXC Chemokine Ligand 4 (CXCL4) Is a Platelet-Derived Mediator of Experimental Liver Fibrosis. Hepatology 2010, 51, 1345–1353. [Google Scholar] [CrossRef]
- Kondo, R.; Yano, H.; Nakashima, O.; Tanikawa, K.; Nomura, Y.; Kage, M. Accumulation of platelets in the liver may be an important contributory factor to thrombocytopenia and liver fibrosis in chronic hepatitis C. J. Gastroenterol. 2013, 48, 526–534. [Google Scholar] [CrossRef]
- Chew, V.; Tow, C.; Teo, M.; Wong, H.L.; Chan, J.; Gehring, A.; Loh, M.; Bolze, A.; Quek, R.; Lee, V.K.M.; et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J. Hepatol. 2010, 52, 370–379. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.C.; Lee, C.H.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323. [Google Scholar] [CrossRef]
- Vasina, E.M.; Cauwenberghs, S.; Feijge, M.A.; Heemskerk, J.W.; Weber, C.; Koenen, R.R. Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death Dis. 2011, 2, e211. [Google Scholar] [CrossRef]
- Vasina, M.; Cauwenberghs, S.; Staudt, M.; Feijge, M.A.H.; Weber, C.; Koenen, R.; Heemskerk, J.W.M. Aging- and activation-induced platelet microparticles suppress apoptosis in monocytic cells and differentially signal to proinflammatory mediator release. Am. J. Blood Res. 2013, 3, 107–123. [Google Scholar]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIb alpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641. [Google Scholar] [CrossRef]
- Pereboom, I.T.A.; Lisman, T.; Porte, R.J. Platelets in liver transplantation: Friend or foe? Liver Transplant. 2008, 14, 923–931. [Google Scholar] [CrossRef]
- Dominguez-Soto, A.; Usategui, A.; de las Casas-Engel, M.; Simon-Fuentes, M.; Nieto, C.; Cuevas, V.D.; Vega, M.A.; Pablos, J.L.; Corbi, A.L. Serotonin drives the acquisition of a profibrotic and anti-inflammatory gene profile through the 5-HT7R-PKA signaling axis. Sci. Rep.-UK 2017, 7, 14761. [Google Scholar] [CrossRef]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Placke, T.; Orgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.L.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Blood 2005, 106, 1167. [Google Scholar] [CrossRef]
- Lang, P.A.; Contaldo, C.; Georgiev, P.; El-Badry, A.M.; Recher, M.; Kurrer, M.; Cervantes-Barragan, L.; Ludewig, B.; Calzascia, T.; Bolinger, B.; et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat. Med. 2008, 14, 756–761. [Google Scholar] [CrossRef]
- Jokisch, J.F.; Grimm, T.; Buchner, A.; Kretschmer, A.; Weinhold, P.; Stief, C.G.; Karl, A.; Schulz, G.B. Preoperative Thrombocytosis in Patients Undergoing Radical Cystectomy for Urothelial Cancer of the Bladder: An Independent Prognostic Parameter for an Impaired Oncological Outcome. Urol. Int. 2019, 2019, 1–6. [Google Scholar] [CrossRef]
- Pedrazzani, C.; Turri, G.; Mantovani, G.; Conti, C.; Ziello, R.; Conci, S.; Campagnaro, T.; Ruzzenente, A.; Guglielmi, A. Prognostic value of thrombocytosis in patients undergoing surgery for colorectal cancer with synchronous liver metastases. Clin. Transl. Oncol. 2019. [Google Scholar] [CrossRef]
- Hwang, S.J.; Luo, J.C.; Li, C.P.; Chu, C.W.; Wu, J.C.; Lai, C.R.; Chiang, J.H.; Chau, G.Y.; Lui, W.Y.; Lee, C.C.; et al. Thrombocytosis: A paraneoplastic syndrome in patients with hepatocellular carcinoma. World J. Gastroenterol. 2004, 10, 2472–2477. [Google Scholar] [CrossRef]
- Nickerson, H.J.; Silberman, T.L.; McDonald, T.P. Hepatoblastoma, thrombocytosis, and increased thrombopoietin. Cancer 1980, 45, 315–317. [Google Scholar] [CrossRef]
- Carr, B.I.; Lin, C.Y.; Lu, S.N. Platelet-related phenotypic patterns in hepatocellular carcinoma patients. Semin. Oncol. 2014, 41, 415–421. [Google Scholar] [CrossRef]
- Zanetto, A.; Senzolo, M.; Vitale, A.; Cillo, U.; Radu, C.; Sartorello, F.; Spiezia, L.; Campello, E.; Rodriguez-Castro, K.; Ferrarese, A.; et al. Thromboelastometry hypercoagulable profiles and portal vein thrombosis in cirrhotic patients with hepatocellular carcinoma. Dig. Liver Dis. 2017, 49, 440–445. [Google Scholar] [CrossRef]
- Carr, B.I.; Guerra, V. Hepatocellular Carcinoma Extrahepatic Metastasis in Relation to Tumor Size and Alkaline Phosphatase Levels. Oncology 2016, 90, 136–142. [Google Scholar] [CrossRef]
- Carr, B.I.; Guerra, V. Thrombocytosis and hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 1790–1796. [Google Scholar] [CrossRef]
- Li, X.; Chen, Z.H.; Xing, Y.F.; Wang, T.T.; Wu, D.H.; Wen, J.Y.; Chen, J.; Lin, Q.; Dong, M.; Wei, L.; et al. Platelet-to-lymphocyte ratio acts as a prognostic factor for patients with advanced hepatocellular carcinoma. Tumor. Biol. 2015, 36, 2263–2269. [Google Scholar] [CrossRef]
- Cho, S.Y.; Yang, J.J.; You, E.; Kim, B.H.; Shim, J.; Lee, H.J.; Lee, W.I.; Suh, J.T.; Park, T.S. Mean platelet volume/platelet count ratio in hepatocellular carcinoma. Platelets 2013, 24, 375–377. [Google Scholar] [CrossRef]
- Li, C.; Wen, T.F.; Yan, L.N.; Li, B.; Wang, W.T.; Yang, J.Y.; Xu, M.Q. Postoperative neutrophil-to-lymphocyte ratio plus platelet-to-lymphocyte ratio predicts the outcomes of hepatocellular carcinoma. J. Surg. Res. 2015, 198, 73–79. [Google Scholar] [CrossRef]
- Pang, Q.; Qu, K.; Zhang, J.Y.; Song, S.D.; Liu, S.S.; Tai, M.H.; Liu, H.C.; Liu, C. The Prognostic Value of Platelet Count in Patients With Hepatocellular Carcinoma A Systematic Review and Meta-Analysis. Medicine 2015, 94. [Google Scholar] [CrossRef]
- Lu, S.N.; Wang, J.H.; Liu, S.L.; Hung, C.H.; Chen, C.H.; Tung, H.D.; Chen, T.M.; Huang, W.S.; Lee, C.M.; Chen, C.C.; et al. Thrombocytopenia as a surrogate for cirrhosis and a marker for the identification of patients at high-risk for hepatocellular carcinoma. Cancer 2006, 107, 2212–2222. [Google Scholar] [CrossRef]
- FDA Approves New Drug for Patients with Chronic Liver Disease Who Have Low Blood Platelets and Are Undergoing A Medical Procedure. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-patients-chronic-liver-disease-who-have-low-blood-platelets-and-are-undergoing (accessed on 4 July 2019).
- Sandler, R.S.; Halabi, S.; Baron, J.A.; Budinger, S.; Paskett, E.; Keresztes, R.; Petrelli, N.; Pipas, J.M.; Karp, D.D.; Loprinzi, C.L.; et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med. 2003, 348, 883–890. [Google Scholar] [CrossRef]
- Simon, T.G.; Ma, Y.; Ludvigsson, J.F.; Chong, D.Q.; Giovannucci, E.L.; Fuchs, C.S.; Meyerhardt, J.A.; Corey, K.E.; Chung, R.T.; Zhang, X.; et al. Association Between Aspirin Use and Risk of Hepatocellular Carcinoma. JAMA Oncol. 2018, 4, 1683–1690. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.Y.; Liu, M.S.; Zhang, C.; Cai, P.C.; Xiao, Z.H.; Wang, F.F. Aspirin enhances the sensitivity of hepatocellular carcinoma side population cells to doxorubicin via miR-491/ABCG2. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Guo, H.; Xu, J.; Li, B.; Liu, Y.J.; Cheng, C.; Zhou, C.; Zhao, Y.; Liu, Y. Activated platelets inhibit hepatocellular carcinoma cell differentiation and promote tumor progression via platelet-tumor cell binding. Oncotarget 2016, 7, 60609–60622. [Google Scholar] [CrossRef]
- Lee, M.; Chung, G.E.; Lee, J.H.; Oh, S.; Nam, J.Y.; Chang, Y.; Cho, H.; Ahn, H.; Cho, Y.Y.; Yoo, J.J.; et al. Antiplatelet therapy and the risk of hepatocellular carcinoma in chronic hepatitis B patients on antiviral treatment. Hepatology 2017, 66, 1556–1569. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, L.A.G.; Soriano-Gabarro, M.; Bromley, S.; Lanas, A.; Soriano, L.C. New use of low-dose aspirin and risk of colorectal cancer by stage at diagnosis: A nested case-control study in UK general practice. BMC Cancer 2017, 17, 637. [Google Scholar] [CrossRef]
- Hwang, I.C.; Chang, J.; Kim, K.; Park, S.M. Aspirin Use and Risk of Hepatocellular Carcinoma in a National Cohort Study of Korean Adults. Sci. Rep. 2018, 8, 4968. [Google Scholar] [CrossRef]
- Lee, T.Y.; Hsu, Y.C.; Tseng, H.C.; Yu, S.H.; Lin, J.T.; Wu, M.S.; Wu, C.Y. Association of Daily Aspirin Therapy With Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. JAMA Intern. Med. 2019. [Google Scholar] [CrossRef]
- Poujol-Robert, A.; Boelle, P.Y.; Conti, F.; Durand, F.; Duvoux, C.; Wendum, D.; Paradis, V.; Mackiewicz, V.; Chazouilleres, O.; Corpechot, C.; et al. Aspirin may reduce liver fibrosis progression: Evidence from a multicenter retrospective study of recurrent hepatitis C after liver transplantation. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 570–576. [Google Scholar] [CrossRef]
- Boas, F.E.; Brown, K.T.; Ziv, E.; Yarmohammadi, H.; Sofocleous, C.T.; Erinjeri, J.P.; Harding, J.J.; Solomon, S.B. Aspirin Is Associated With Improved Liver Function After Embolization of Hepatocellular Carcinoma. AJR Am. J. Roentgenol. 2019, 1–7. [Google Scholar] [CrossRef]
- Li, J.H.; Wang, Y.; Xie, X.Y.; Yin, X.; Zhang, L.; Chen, R.X.; Ren, Z.G. Aspirin in combination with TACE in treatment of unresectable HCC: A matched-pairs analysis. Am. J. Cancer Res. 2016, 6, 2109–2116. [Google Scholar]
- Liu, Y.X.; Feng, J.Y.; Sun, M.M.; Liu, B.W.; Yang, G.; Bu, Y.N.; Zhao, M.; Wang, T.J.; Zhang, W.Y.; Yuan, H.F.; et al. Aspirin inhibits the proliferation of hepatoma cells through controlling GLUT1-mediated glucose metabolism. Acta Pharmacol. Sin. 2019, 40, 122–132. [Google Scholar] [CrossRef]
- Li, S.; Dai, W.; Mo, W.; Li, J.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int. J. Cancer 2017, 141, 2571–2584. [Google Scholar] [CrossRef]
- Hossain, M.A.; Kim, D.H.; Jang, J.Y.; Kang, Y.J.; Yoon, J.H.; Moon, J.O.; Chung, H.Y.; Kim, G.Y.; Choi, Y.H.; Copple, B.L.; et al. Aspirin enhances doxorubicin-induced apoptosis and reduces tumor growth in human hepatocellular carcinoma cells in vitro and in vivo. Int. J. Oncol. 2012, 40, 1636–1642. [Google Scholar] [CrossRef]
- Hossain, M.A.; Kim, D.H.; Jang, J.Y.; Kang, Y.J.; Yoon, J.H.; Moon, J.O.; Chung, H.Y.; Kim, G.Y.; Choi, Y.H.; Copple, B.L.; et al. Aspirin induces apoptosis in vitro and inhibits tumor growth of human hepatocellular carcinoma cells in a nude mouse xenograft model. Int. J. Oncol. 2012, 40, 1298–1304. [Google Scholar] [CrossRef]
- Lee, P.C.; Yeh, C.M.; Hu, Y.W.; Chen, C.C.; Liu, C.J.; Su, C.W.; Huo, T.I.; Huang, Y.H.; Chao, Y.; Chen, T.J.; et al. Antiplatelet Therapy is Associated with a Better Prognosis for Patients with Hepatitis B Virus-Related Hepatocellular Carcinoma after Liver Resection. Ann. Surg. Oncol. 2016, 23, S874–S883. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Brit. J. Clin. Pharmacol. 2011, 72, 634–646. [Google Scholar] [CrossRef]
- Akcan, A.; Kucuk, C.; Ok, E.; Canoz, O.; Muhtaroglu, S.; Yilmaz, N.; Yilmaz, Z. The effect of amrinone on liver regeneration in experimental hepatic resection model. J. Surg. Res. 2006, 130, 66–72. [Google Scholar] [CrossRef]
- Gresele, P.; Deckmyn, H.; Nenci, G.G.; Vermylen, J. Thromboxane Synthase Inhibitors, Thromboxane Receptor Antagonists and Dual Blockers in Thrombotic Disorders. Trends Pharmacol. Sci. 1991, 12, 158–163. [Google Scholar] [CrossRef]
- Yokoyama, I.; Hayashi, S.; Kobayashi, T.; Negita, M.; Yasutomi, M.; Uchida, K.; Takagi, H. Prevention of Experimental Hepatic Metastasis with Thromboxane Synthase Inhibitor. Res. Exp. Med. 1995, 195, 209–215. [Google Scholar] [CrossRef]
- Nanji, A.A.; Liong, E.C.; Xiao, J.; Tipoe, G.L. Thromboxane inhibitors attenuate inflammatory and fibrotic changes in rat liver despite continued ethanol administrations. Alcohol. Clin. Exp. Res. 2013, 37, 31–39. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlovic, N.; Rani, B.; Gerwins, P.; Heindryckx, F. Platelets as Key Factors in Hepatocellular Carcinoma. Cancers 2019, 11, 1022. https://doi.org/10.3390/cancers11071022
Pavlovic N, Rani B, Gerwins P, Heindryckx F. Platelets as Key Factors in Hepatocellular Carcinoma. Cancers. 2019; 11(7):1022. https://doi.org/10.3390/cancers11071022
Chicago/Turabian StylePavlovic, Natasa, Bhavna Rani, Pär Gerwins, and Femke Heindryckx. 2019. "Platelets as Key Factors in Hepatocellular Carcinoma" Cancers 11, no. 7: 1022. https://doi.org/10.3390/cancers11071022
APA StylePavlovic, N., Rani, B., Gerwins, P., & Heindryckx, F. (2019). Platelets as Key Factors in Hepatocellular Carcinoma. Cancers, 11(7), 1022. https://doi.org/10.3390/cancers11071022