Flavopereirine Suppresses the Growth of Colorectal Cancer Cells through P53 Signaling Dependence

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Flavopereirine Reduces the Viability of Certain CRC Cell Lines
2.2. Flavopereirine Promotes Intrinsic and Extrinsic Apoptosis in CRC Cells
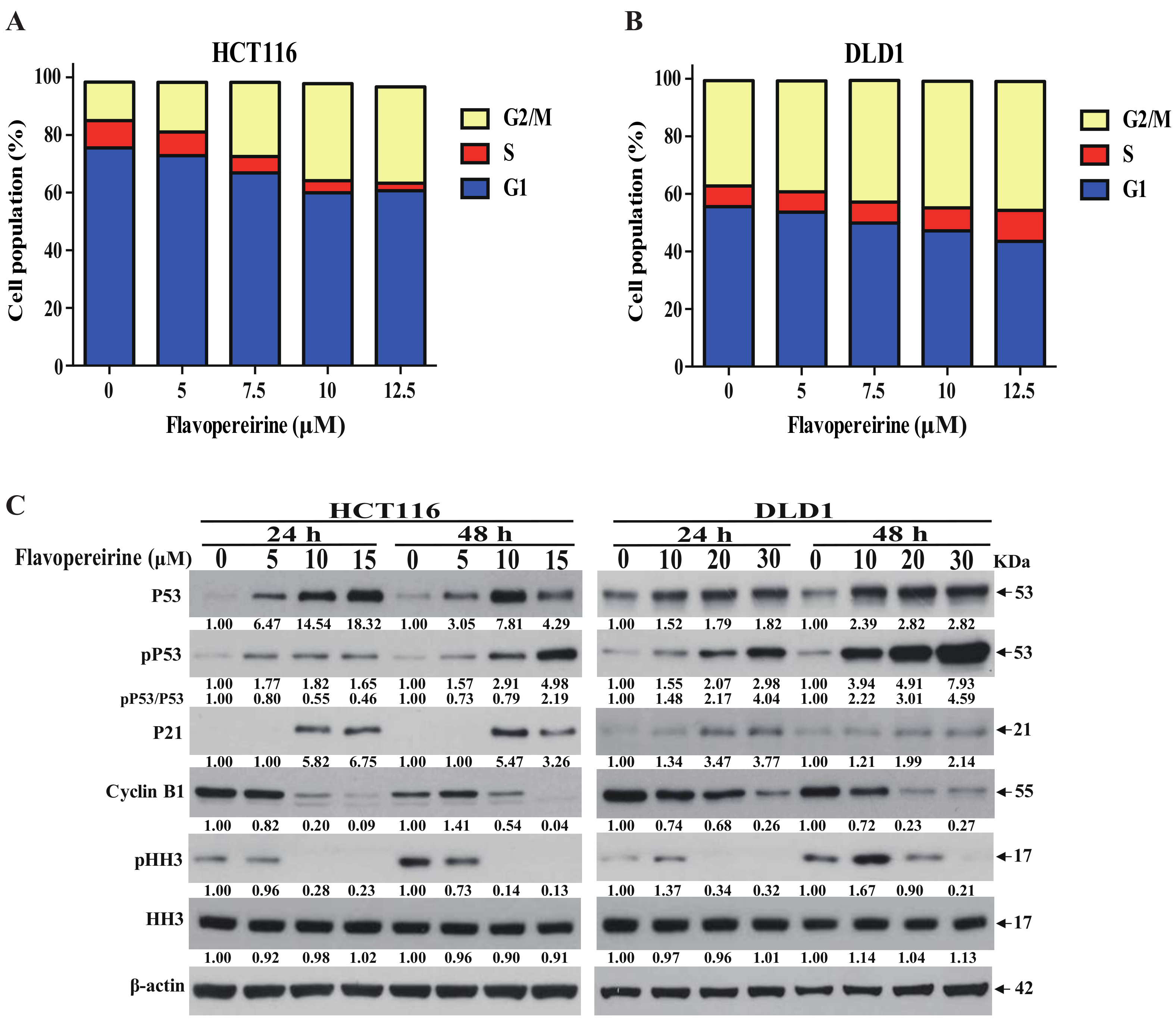
2.3. Flavopereirine Stimulates G2/M-Phase Cell Cycle Arrest in CRC Cells
2.4. Flavopereirine Lowers CRC Cell Viability via JAKs-STATs-c-Myc Signaling but Is Not Dependent on It
2.5. P53 Signaling Is Involved in Flavopereirine-Mediated Viability Reduction and Apoptosis Induction in CRC Cells
2.6. Flavopereirine Significantly Inhibits HCT116-Xenograft Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Flavopereirine
4.3. Viability Assay
4.4. Apoptosis Assay
4.5. Western Blotting Analysis
4.6. Cell Cycle Analysis
4.7. Enforced Expression of Constitutive Active STAT3 (cSTAT3) and c-Myc in CRC Cells
4.8. In Vivo Tumor Growth Assay
4.9. Immunohistochemical and Immunofluorescent TUNEL Staining Assays of HCT116-Xenograft Tumor
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Favoriti, P.; Carbone, G.; Greco, M.; Pirozzi, F.; Pirozzi, R.E.; Corcione, F. Worldwide burden of colorectal cancer: A review. Updates Surg. 2016, 68, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, X.; Feng, W.; Guo, H.; Tang, C.; Lu, Y.; Xiang, X.; Bao, Y. Gene signatures associated with drug resistance to irinotecan and oxaliplatin predict a poor prognosis in patients with colorectal cancer. Oncol. Lett. 2017, 13, 2089–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers 2013, 5, 676–713. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Schindler, C.W. Series introduction. JAK-STAT signaling in human disease. J. Clin. Investig. 2002, 109, 1133–1137. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. The Stat family in cytokine signaling. Curr. Opin. Cell Biol. 2001, 13, 211–217. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zushi, S.; Shinomura, Y.; Kiyohara, T.; Miyazaki, Y.; Kondo, S.; Sugimachi, M.; Higashimoto, Y.; Kanayama, S.; Matsuzawa, Y. STAT3 mediates the survival signal in oncogenic ras-transfected intestinal epithelial cells. Int. J. Cancer 1998, 78, 326–330. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitzner, M.; Roesler, B.; Bielfeld, C.; Emons, G.; Gaedcke, J.; Wolff, H.A.; Rave-Frank, M.; Kramer, F.; Beissbarth, T.; Kitz, J.; et al. STAT3 inhibition sensitizes colorectal cancer to chemoradiotherapy in vitro and in vivo. Int. J. Cancer 2014, 134, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar] [CrossRef]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. Regulation of p53 downstream genes. Semin. Cancer Biol. 1998, 8, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Robles, A.I.; Jen, J.; Harris, C.C. Clinical Outcomes of TP53 Mutations in Cancers. Cold Spring Harb Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Wiegering, A.; Matthes, N.; Muhling, B.; Koospal, M.; Quenzer, A.; Peter, S.; Germer, C.T.; Linnebacher, M.; Otto, C. Reactivating p53 and Inducing Tumor Apoptosis (RITA) Enhances the Response of RITA-Sensitive Colorectal Cancer Cells to Chemotherapeutic Agents 5-Fluorouracil and Oxaliplatin. Neoplasia 2017, 19, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Hughes, N.A.; Rapoport, H. Flavopereirine, an Alkaloids from Geissospermum vellosii. J. Am. Chem. Soc. 1958, 80, 1604–1609. [Google Scholar] [CrossRef]
- Beljanski, M.; Beljanski, M.S. Selective inhibition of in vitro synthesis of cancer DNA by alkaloids of beta-carboline class. Exp. Cell Biol. 1982, 50, 79–87. [Google Scholar] [CrossRef]
- Beljanski, M. The anticancer agent PB-100, selective active on malignant cells, inhibits multiplication of sixteen malignant cell lines, evenmultidrug resistant. Genet. Mol. Biol. 2000, 20, 29–33. [Google Scholar] [CrossRef]
- Beljanski, M.; Crochet, S.; Beljanski, M.S. PB-100: A potent and selective inhibitor of human BCNU resistant glioblastoma cell multiplication. Anticancer Res. 1993, 13, 2301–2308. [Google Scholar] [PubMed]
- Beljanski, M.; Crochet, S. The selective anticancer agent pb-100 inhibits interleukin-6 induced enhancement of glioblastoma cell-proliferation in-vitro. Int. J. Oncol. 1994, 5, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Innocente, S.A.; Abrahamson, J.L.; Cogswell, J.P.; Lee, J.M. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. USA 1999, 96, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Hans, F.; Dimitrov, S. Histone H3 phosphorylation and cell division. Oncogene 2001, 20, 3021–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.W.; Harris, C.C. p53 tumor-suppressor gene: Clues to molecular carcinogenesis. J. Cell Physiol. 1997, 173, 247–255. [Google Scholar] [CrossRef]
- Djelloul, S.; Forgue-Lafitte, M.E.; Hermelin, B.; Mareel, M.; Bruyneel, E.; Baldi, A.; Giordano, A.; Chastre, E.; Gespach, C. Enterocyte differentiation is compatible with SV40 large T expression and loss of p53 function in human colonic Caco-2 cells. Status of the pRb1 and pRb2 tumor suppressor gene products. FEBS Lett. 1997, 406, 234–242. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Baba, K.; Oshita, A.; Kohyama, M.; Inoue, S.; Kuroo, Y.; Yamaguchi, T.; Nakamura, H.; Sugiyama, Y.; Tazaki, T.; Sasaki, M.; et al. Successful treatment of conversion chemotherapy for initially unresectable synchronous colorectal liver metastasis. World J. Gastroenterol. 2015, 21, 1982–1988. [Google Scholar] [CrossRef]
- Chan, K.M.; Wu, T.H.; Cheng, C.H.; Lee, W.C.; Chiang, J.M.; Chen, J.S.; Wang, J.Y. Prognostic significance of the number of tumors and aggressive surgical approach in colorectal cancer hepatic metastasis. World J. Surg. Oncol. 2014, 12, 155. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.Y.; Yang, X.F.; Shen, D.F.; Sun, H.Z.; Huang, K.Q.; Zheng, H.C. Effects and mechanism of STAT3 silencing on the growth and apoptosis of colorectal cancer cells. Oncol. Lett. 2018, 16, 5575–5582. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Ruppert, J.M.; Aster, J.C.; Winchester, E. Inhibition of DNA replication factor RPA by p53. Nature 1993, 365, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- Saldana-Meyer, R.; Recillas-Targa, F. Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 2011, 6, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazan-Mamczarz, K.; Galban, S.; Lopez de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffee, P.; Osipov, A.; Tan, C.; Tuli, R.; Hendifar, A. Review of systemic therapies for locally advanced and metastatic rectal cancer. J. Gastrointest. Oncol. 2015, 6, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Boyer, J.; Allen, W.L.; Latif, T.; Ferguson, P.R.; Maxwell, P.J.; McDermott, U.; Lynch, M.; Harkin, D.P.; Johnston, P.G. The role of thymidylate synthase induction in modulating p53-regulated gene expression in response to 5-fluorouracil and antifolates. Cancer Res. 2002, 62, 2644–2649. [Google Scholar] [PubMed]
- Skinner, H.D.; Sandulache, V.C.; Ow, T.J.; Meyn, R.E.; Yordy, J.S.; Beadle, B.M.; Fitzgerald, A.L.; Giri, U.; Ang, K.K.; Myers, J.N. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin. Cancer Res. 2012, 18, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Grinkevich, V.V.; Nikulenkov, F.; Shi, Y.; Enge, M.; Bao, W.; Maljukova, A.; Gluch, A.; Kel, A.; Sangfelt, O.; Selivanova, G. Ablation of key oncogenic pathways by RITA-reactivated p53 is required for efficient apoptosis. Cancer Cell 2009, 15, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Wheeler, D.L. KRAS mutant colorectal tumors: Past and present. Small GTPases 2012, 3, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.D.; Piperdi, B. KRAS mutation in colon cancer: A marker of resistance to EGFR-I therapy. Ann. Surg. Oncol. 2010, 17, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, K.; Zhang, L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015, 2, 4–12. [Google Scholar] [CrossRef]
- Lai, H.; Wang, Y.; Duan, F.; Li, Y.; Jiang, Z.; Luo, L.; Liu, L.; Leung, E.L.H.; Yao, X. Krukovine Suppresses KRAS-Mutated Lung Cancer Cell Growth and Proliferation by Inhibiting the RAF-ERK Pathway and Inactivating AKT Pathway. Front. Pharmacol. 2018, 9, 958. [Google Scholar] [CrossRef]
- Boyer, J.; McLean, E.G.; Aroori, S.; Wilson, P.; McCulla, A.; Carey, P.D.; Longley, D.B.; Johnston, P.G. Characterization of p53 wild-type and null isogenic colorectal cancer cell lines resistant to 5-fluorouracil, oxaliplatin, and irinotecan. Clin. Cancer Res. 2004, 10, 2158–2167. [Google Scholar] [CrossRef]
- Shi, C.S.; Huang, H.C.; Wu, H.L.; Kuo, C.H.; Chang, B.I.; Shiao, M.S.; Shi, G.Y. Salvianolic acid B modulates hemostasis properties of human umbilical vein endothelial cells. Thromb. Res. 2007, 119, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Doshi, U.A.; Shaw, J.; Fox, T.E.; Claxton, D.F.; Loughran, T.P.; Kester, M. STAT3 mediates C6-ceramide-induced cell death in chronic lymphocytic leukemia. Signal Transduct. Target. Ther. 2017, 2, 17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Gonzalez, J.M.; Armaiz-Pena, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarria-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejia, P.E. Targeting c-MYC in Platinum-Resistant Ovarian Cancer. Mol. Cancer Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.S.; Jou, Y.C.; Lee, G.F.; Chen, Y.C.; Shiau, A.L.; Tsai, H.T.; Wu, C.L.; Tzai, T.S. Aberrant prothymosin-alpha expression in human bladder cancer. Urology 2009, 73, 188–192. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.-M.; Huang, Y.-C.; Kuo, Y.-H.; Cheng, C.-C.; Kuan, F.-C.; Chang, S.-F.; Lee, Y.-R.; Chin, C.-C.; Shi, C.-S. Flavopereirine Suppresses the Growth of Colorectal Cancer Cells through P53 Signaling Dependence. Cancers 2019, 11, 1034. https://doi.org/10.3390/cancers11071034
Li J-M, Huang Y-C, Kuo Y-H, Cheng C-C, Kuan F-C, Chang S-F, Lee Y-R, Chin C-C, Shi C-S. Flavopereirine Suppresses the Growth of Colorectal Cancer Cells through P53 Signaling Dependence. Cancers. 2019; 11(7):1034. https://doi.org/10.3390/cancers11071034
Chicago/Turabian StyleLi, Jhy-Ming, Yun-Ching Huang, Yi-Hung Kuo, Chih-Chung Cheng, Feng-Che Kuan, Shun-Fu Chang, Ying-Ray Lee, Chih-Chien Chin, and Chung-Sheng Shi. 2019. "Flavopereirine Suppresses the Growth of Colorectal Cancer Cells through P53 Signaling Dependence" Cancers 11, no. 7: 1034. https://doi.org/10.3390/cancers11071034
APA StyleLi, J. -M., Huang, Y. -C., Kuo, Y. -H., Cheng, C. -C., Kuan, F. -C., Chang, S. -F., Lee, Y. -R., Chin, C. -C., & Shi, C. -S. (2019). Flavopereirine Suppresses the Growth of Colorectal Cancer Cells through P53 Signaling Dependence. Cancers, 11(7), 1034. https://doi.org/10.3390/cancers11071034