A Novel AURKA Mutant-Induced Early-Onset Severe Hepatocarcinogenesis Greater than Wild-Type via Activating Different Pathways in Zebrafish

 ,
,
Abstract
:1. Introduction
2. Results
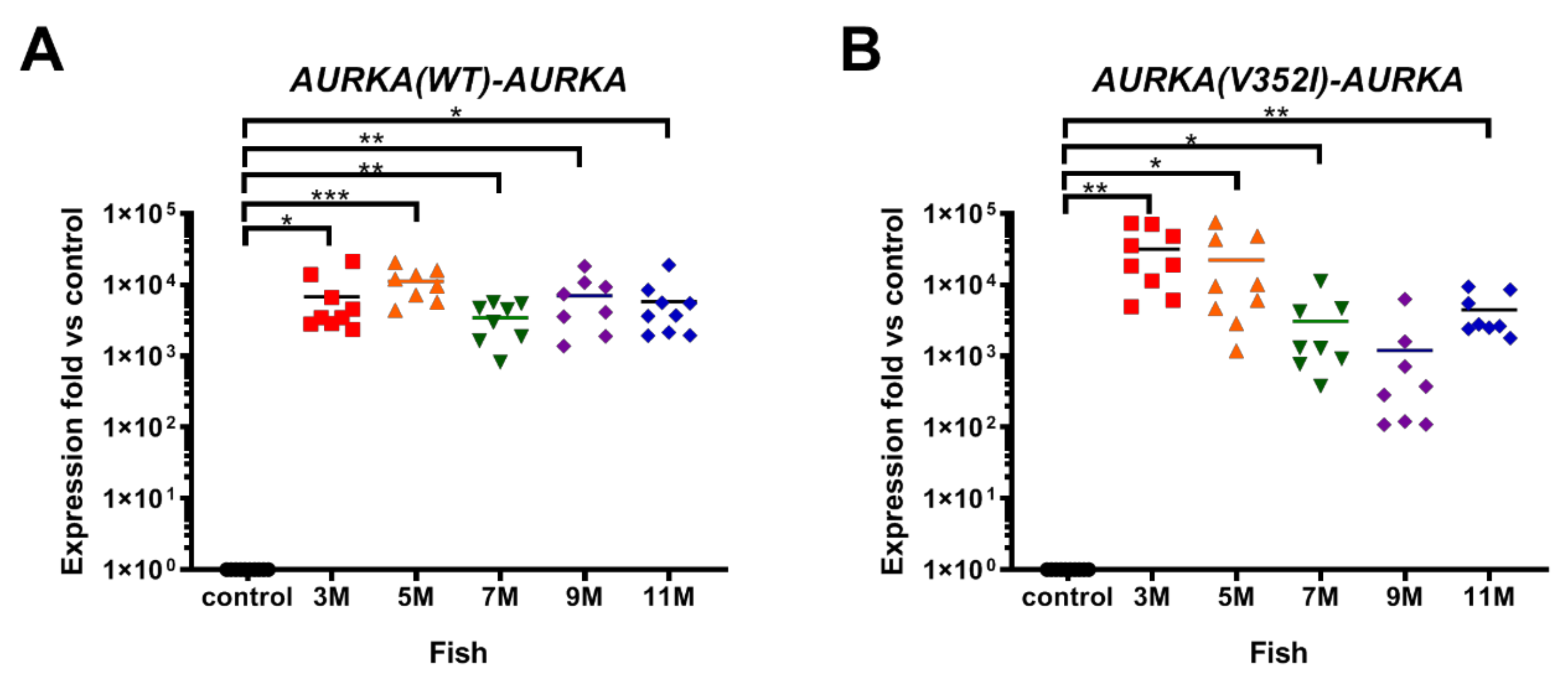
2.1. Overexpression of AURKA is Dramatically Increased in Transgenic Zebrafish Compared to the Control Zebrafish
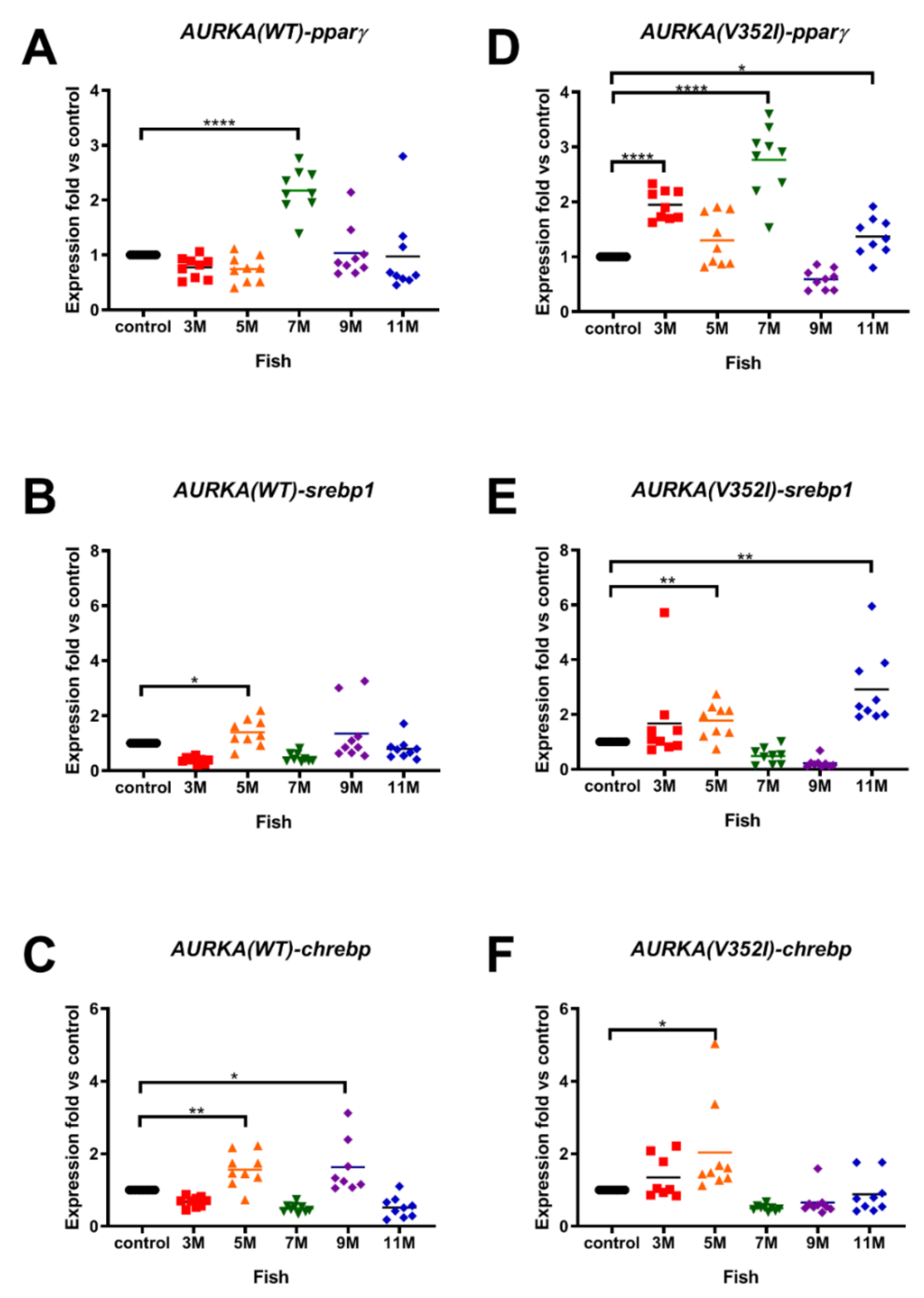
2.2. The Expression of Lipogenic Enzyme and Lipogenic Factor Were Observed in Both AURKA(V352I) and AURKA(WT) Transgenic Fish
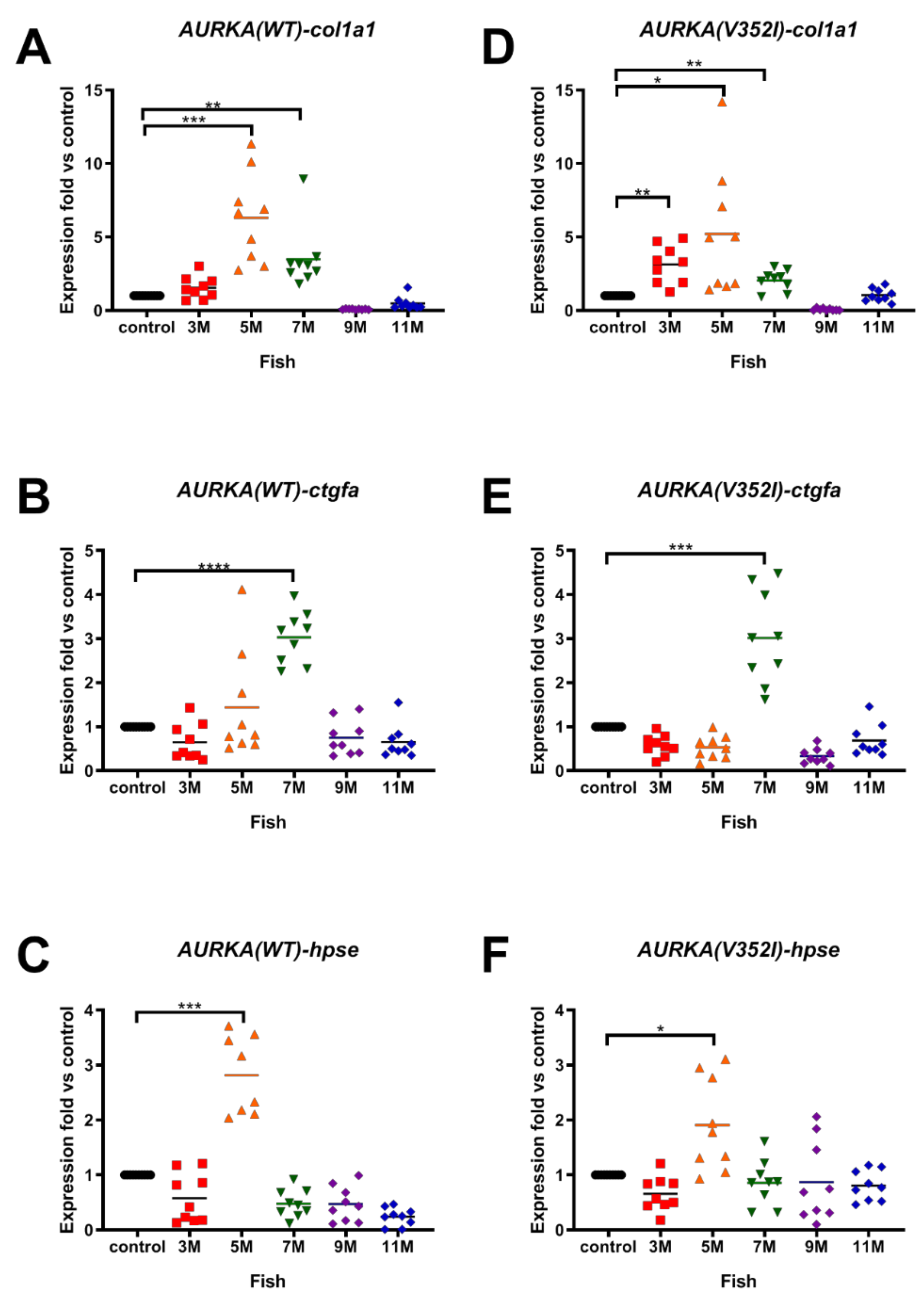
2.3. AURKA(V352I) Has More Probability in Promoting Fibrosis at 3 Months
2.4. Expression of Cell Cycle-Related Genes/Proliferation Markers Were Significantly much Higher and Earlier in AURKA(V352I) than in AURKA(WT) Transgenic Zebrafish
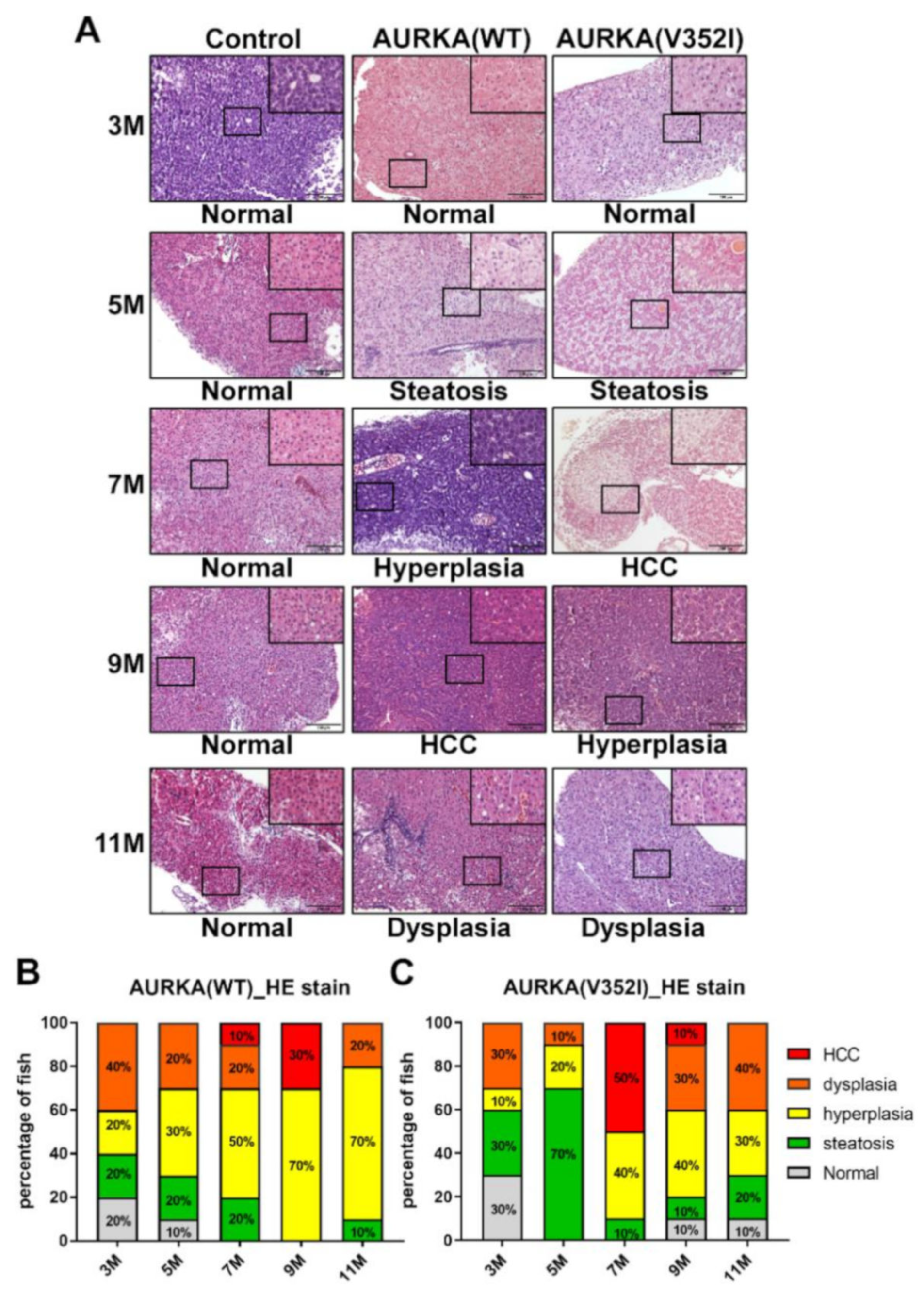
2.5. Hematoxylin and Eosin Staining Reveals that AURKA(V352I) Promotes HCC at 7 Months, Whereas AURKA(WT) Promotes HCC Later at 9 Months
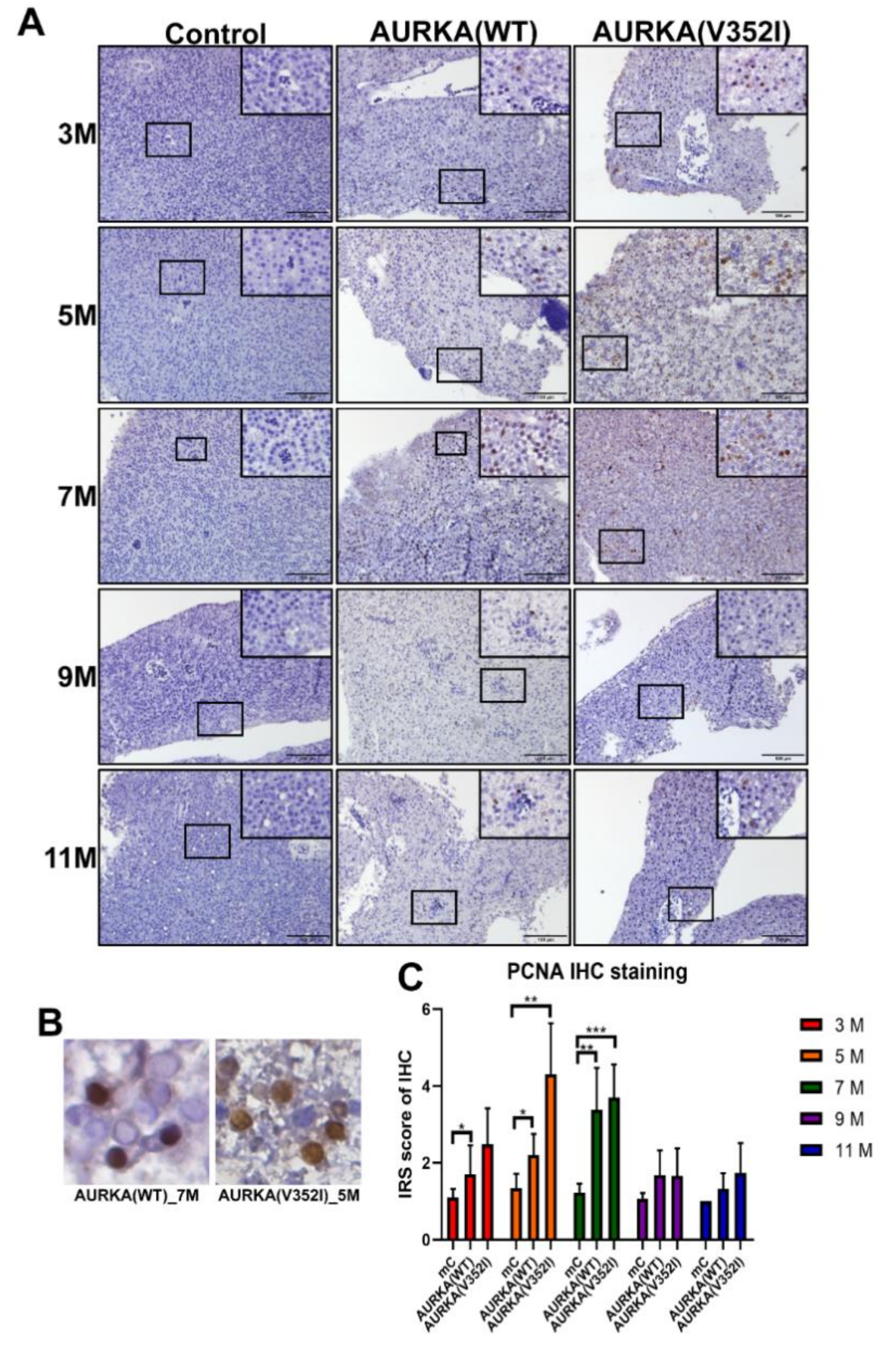
2.6. Proliferating Cell Nuclear Antigen Staining Indicated that Cell Proliferation in AURKA(V352I) Was More Severe than AURKA(WT) and Control Fish from 3 to 7 Months
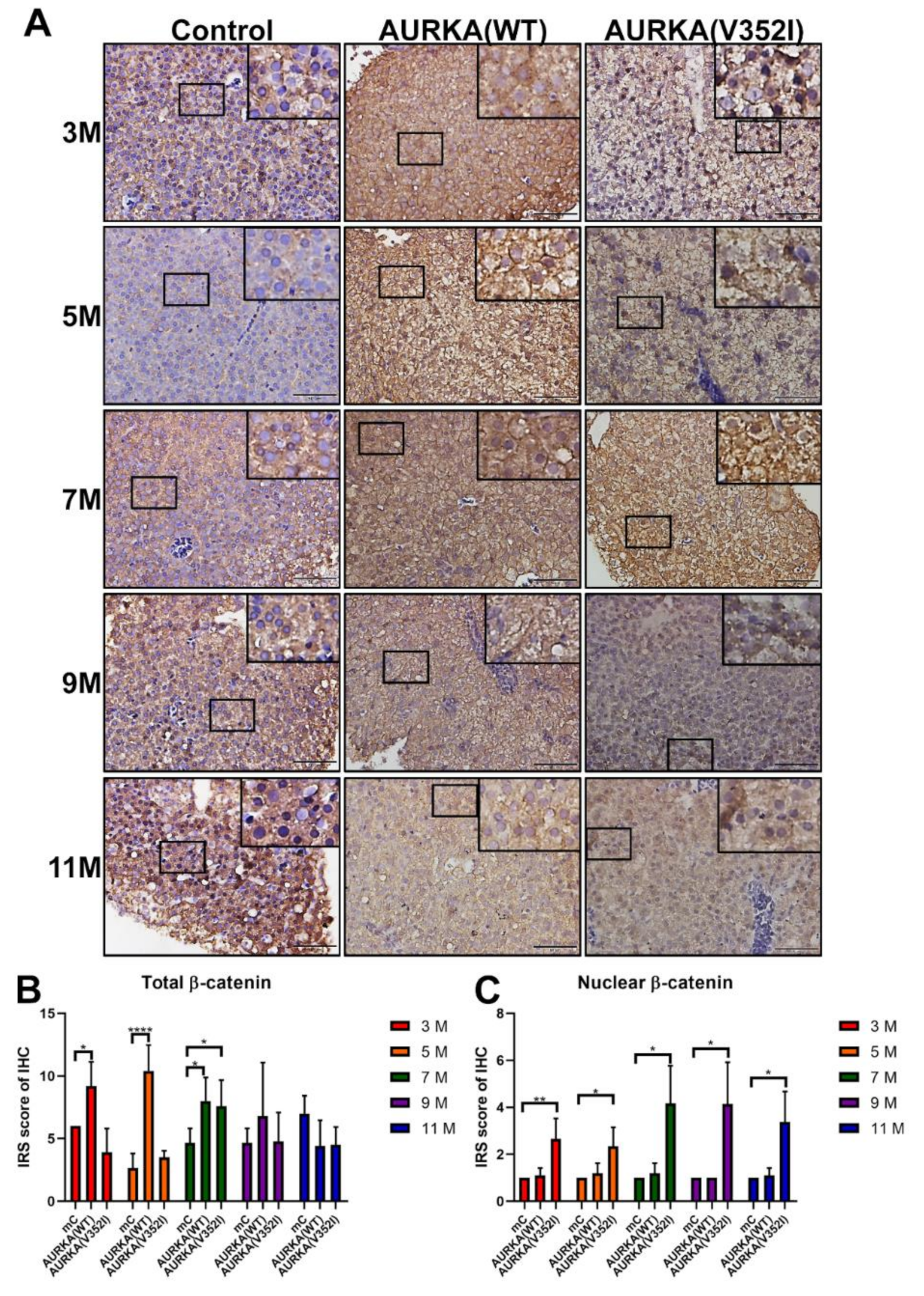
2.7. Less Membrane Bound β-Catenin for Metastatic Behavior of Hepatocyte in AURKA(V352I) Transgenic Fish for EMT Transition
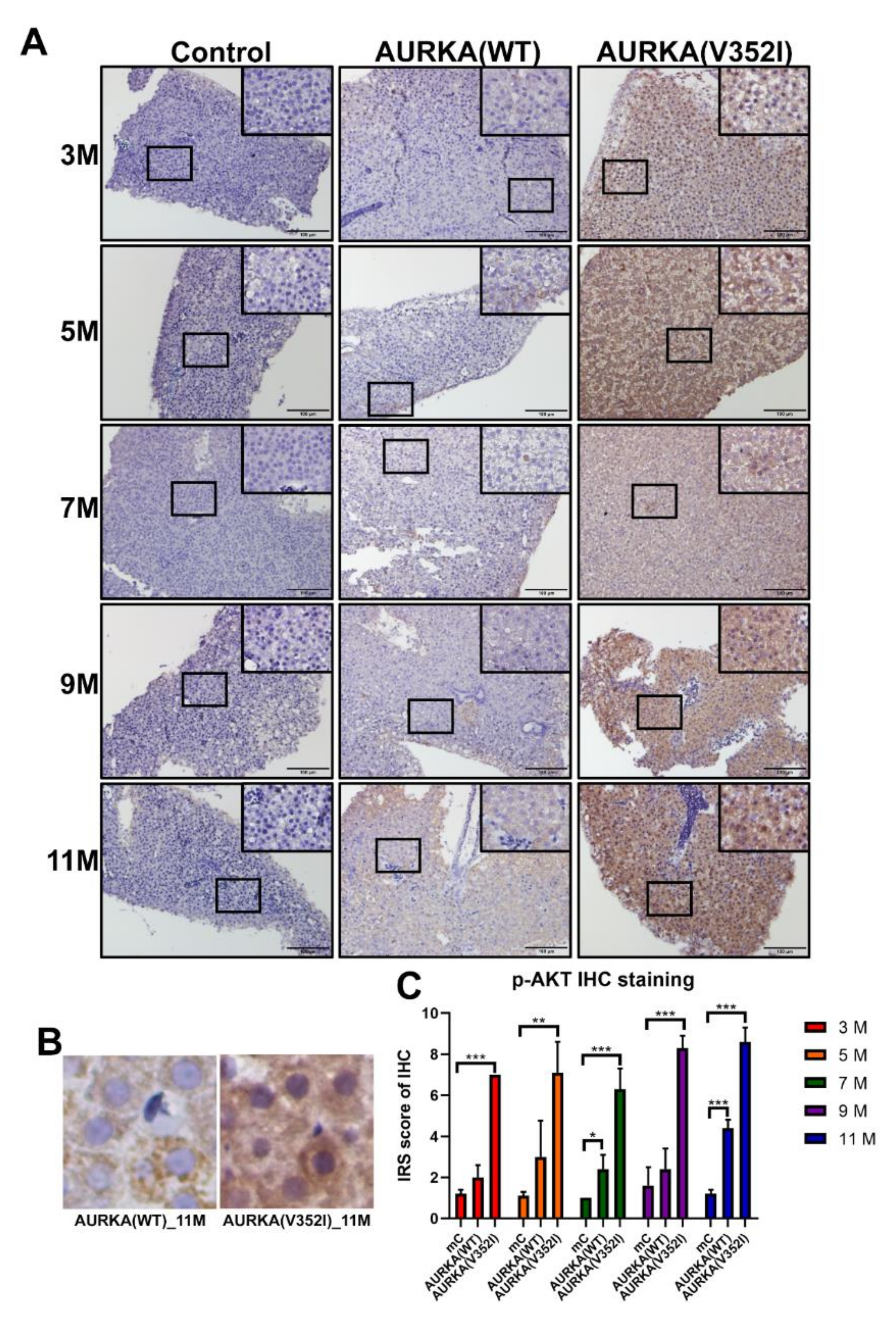
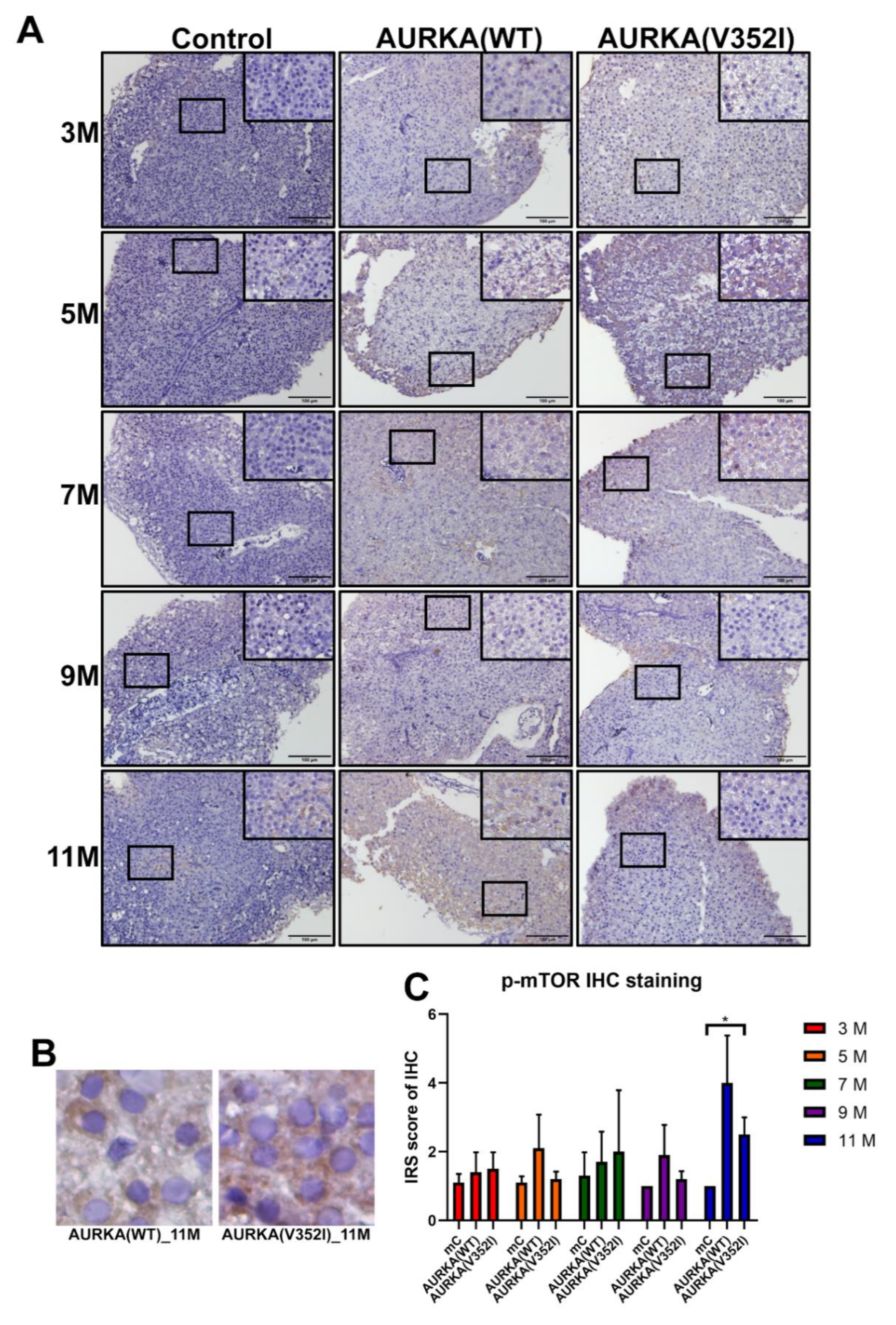
2.8. The pAKT in AURKA(V352I) Was Significantly Higher than AURKA(WT) and Control Fish in all Stages
2.9. Multiple Kinase Inhibitors Cannot Prevent Cancer Formation in AURKA(WT) and AURKA(V352I) Transgenic Fish
3. Discussion
4. Materials and Methods
4.1. Generate Tol2 Expression Constructs
4.2. Zebrafish Maintenance
4.3. Liver Tissue Collection and Paraffin Section
4.4. Hematoxylin and Eosin Staining
4.5. Immunohistochemistry Staining
4.6. Total RNA Isolation
4.7. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.8. Quantitative Polymerase Chain Reaction (qPCR)
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roberts, L.R.; Gores, G.J. Hepatocellular carcinoma: Molecular pathways and new therapeutic targets. Semin. Liver Dis. 2005, 25, 212–225. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Samonakis, D.N.; Kouroumalis, E.A. Systemic treatment for hepatocellular carcinoma: Still unmet expectations. World J. Hepatol. 2017, 9, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, I.A.; Tsoulfas, G. Evolving role of Sorafenib in the management of hepatocellular carcinoma. World J. Clin. Oncol. 2017, 8, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, J.; Shen, X. Genome-wide association study: New genetic insights into HBV/HCV-related hepatocellular carcinoma genomes. Scand. J. Gastroenterol 2017, 52, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Spitsbergen, J.M.; Kent, M.L. The state of the art of the zebrafish model for toxicology and toxicologic pathology research—Advantages and current limitations. Toxicol. Pathol. 2003, 31, 62–87. [Google Scholar]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Huiting, L.N.; Laroche, F.; Feng, H. The Zebrafish as a Tool to Cancer Drug Discovery. Austin J. Pharmacol. Ther. 2015, 3, 1069. [Google Scholar] [PubMed]
- Kirchberger, S.; Sturtzel, C.; Pascoal, S.; Distel, M. Quo natas, Danio?-Recent Progress in Modeling Cancer in Zebrafish. Front. Oncol. 2017, 7, 186. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.A. Zebrafish and the Cancer Moonshot. Zebrafish 2017, 14, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Yang, W.Y.; Lin, Y.M.; Jin, S.L.; Yuh, C.H. Hepatitis B virus X antigen and aflatoxin B1 synergistically cause hepatitis, steatosis and liver hyperplasia in transgenic zebrafish. Acta Histochem. 2013, 115, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Liao, C.Y.; Yang, W.Y.; Lin, Y.M.; Jin, S.L.; Wang, H.D.; Yuh, C.H. Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PLoS ONE 2014, 9, e85318. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.C.; Hsiao, H.Y.; Tu, H.C.; Yang, W.Y.; Tsai, S.L.; Lin, H.K.; Liao, C.Y.; Lu, J.W.; Chou, Y.T.; Wang, H.D.; et al. Up-Regulation of Golgi α-Mannosidase IA and Down-Regulation of Golgi α-Mannosidase IC Activates Unfolded Protein Response During Hepatocarcinogenesis. Hepatol. Commun. 2017, 1, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.T.; Jiang, J.K.; Yang, M.H.; Lu, J.W.; Lin, H.K.; Wang, H.D.; Yuh, C.H. Identification of a noncanonical function for ribose-5-phosphate isomerase A promotes colorectal cancer formation by stabilizing and activating beta-catenin via a novel C-terminal domain. PLoS Biol. 2018, 16, e2003714. [Google Scholar] [CrossRef]
- Chou, Y.T.; Chen, L.Y.; Tsai, S.L.; Tu, H.C.; Lu, J.W.; Ciou, S.C.; Wang, H.D.; Yuh, C.H. Ribose-5-Phosphate Isomerase A Overexpression Promotes Liver Cancer Development in Transgenic Zebrafish via Activation of ERK and beta-catenin Pathways. Carcinogenesis 2018. [Google Scholar] [CrossRef]
- Kollareddy, M.; Dzubak, P.; Zheleva, D.; Hajduch, M. Aurora kinases: Structure, functions and their association with cancer. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc 2008, 152, 27–33. [Google Scholar] [CrossRef]
- Giet, R.; Prigent, C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine—Threonine kinases. J. Cell Sci. 1999, 3601, 3591–3601. [Google Scholar]
- Carmena, M.; Earnshaw, W.C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Roghi, C.; Giet, R.; Uzbekov, R.; Morin, N.; Chartrain, I.; Guellec, R.L.; Couturier, A.; Dorée, M.; Philippe, M.; Prigent, C. The Xenopus protein kinase pEg2 associates with the centrosome in a cell cycle-dependent manner, binds to the spindle microtubules and is involved in bipolar mitotic spindle assembly. J. Cell Sci. 1998, 572, 557–572. [Google Scholar]
- Giet, R.; Prigent, C. The Xenopus laevis aurora/Ip11p-related kinase pEg2 participates in the stability of the bipolar mitotic spindle. Exp. Cell Res. 2000, 258, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.B.; Salmon, E.D. Chromosome Dynamics: New Light on Aurora B Kinase Function Aurora B family kinases play an essential role in. Curr. Biol. 2002, 12, 458–460. [Google Scholar] [CrossRef]
- Castro, A.; Vigneron, S.; Bernis, C.; Labbé, J.-c.; Prigent, C.; Lorca, T. The D-Box-activating domain (DAD) is a new proteolysis signal that stimulates the silent D-Box sequence of Aurora-A. EMBO Rep. 2002, 3, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A Novel Mechanism for Activation of the Protein Kinase Aurora A. Curr. Biol. 2003, 13, 691–697. [Google Scholar] [CrossRef]
- Fu, J.; Bian, M.; Jiang, Q.; Zhang, C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 2007, 5, 1–10. [Google Scholar] [CrossRef]
- D’Assoro, A.B.; Haddad, T.; Galanis, E. Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front. Oncol. 2015, 5, 295. [Google Scholar] [CrossRef]
- Tsunematsu, T.; Arakaki, R.; Yamada, A.; Ishimaru, N.; Kudo, Y. The Non-Canonical Role of Aurora-A in DNA Replication. Front. Oncol. 2015, 5, 187. [Google Scholar] [CrossRef] [Green Version]
- Jeng, Y.-M.; Peng, S.-Y.; Lin, C.-Y.; Hsu, H.-C. Overexpression and Amplification of Aurora-A in Hepatocellular Carcinoma. Clin. Cancer Res. 2004, 10, 2065–2071. [Google Scholar] [CrossRef]
- Chen, C.; Song, G.; Xiang, J.; Zhang, H.; Zhao, S.; Zhan, Y. AURKA promotes cancer metastasis by regulating epithelial-mesenchymal transition and cancer stem cell properties in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 486, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Yang, N.K.; Liu, T.Y.; Tai, S.K.; Hsu, D.S.; Chen, Y.W.; Chen, Y.J.; Chang, C.C.; Tzeng, C.H.; Yang, M.H. Chromosome instability modulated by BMI1-AURKA signaling drives progression in head and neck cancer. Cancer Res. 2013, 73, 953–966. [Google Scholar] [CrossRef] [PubMed]
- D’Assoro, A.B.; Liu, T.; Quatraro, C.; Amato, A.; Opyrchal, M.; Leontovich, A.; Ikeda, Y.; Ohmine, S.; Lingle, W.; Suman, V.; et al. The mitotic kinase Aurora—A promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERalpha(+) breast cancer cells. Oncogene 2014, 33, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Penrhyn-Lowe, S.; Venkitaraman, A.R. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 2003, 3, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Li, C.C.; Chu, H.Y.; Yang, C.W.; Chou, C.K.; Tsai, T.F. Aurora-A overexpression in mouse liver causes p53-dependent premitotic arrest during liver regeneration. Mol. Cancer Res. 2009, 7, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Lu, L.; Liu, X.; Guo, B.; Zhai, Y.; Li, Y.; Wang, Y.; Xie, B.; Ren, Q.; Cao, P.; et al. Association between the functional polymorphism Ile31Phe in the AURKA gene and susceptibility of hepatocellular carcinoma in chronic hepatitis B virus carriers. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Song, A.P.; Wang, H.; Xie, Y.T.; Han, J.Y.; Sajdik, C.; Tian, X.X.; Fang, W.G. Genetic polymorphisms in AURKA and BRCA1 are associated with breast cancer susceptibility in a Chinese Han population. J. Pathol. 2011, 225, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Song, A.P.; Chen, L.; Liu, D.G.; Li, X.H.; Guo, H.Y.; Tian, X.X.; Fang, W.G. Association of genetic polymorphisms in AURKA, BRCA1, CCNE1 and CDK2 with the risk of endometrial carcinoma and clinicopathological parameters among Chinese Han women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2015, 184, 65–72. [Google Scholar] [CrossRef]
- Chou, C.H.; Chou, Y.E.; Chuang, C.Y.; Yang, S.F.; Lin, C.W. Combined effect of genetic polymorphisms of AURKA and environmental factors on oral cancer development in Taiwan. PLoS ONE 2017, 12, e0171583. [Google Scholar] [CrossRef]
- Pickhard, A.; Siegl, M.; Baumann, A.; Huhn, M.; Wirth, M.; Reiter, R.; Rudelius, M.; Piontek, G.; Brockhoff, G. The response of head and neck squamous cell carcinoma to cetuximab treatment depends on Aurora kinase A polymorphism. Oncotarget 2014, 5, 5428–5438. [Google Scholar] [CrossRef]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell Cycle Control by PTEN. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Yeomanson, D.; Bryant, H.E. PI3King the lock: Targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediatr. Hematol. Oncol. 2015, 37, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A.; Yellen, P.; Xu, L.; Saqcena, M. Regulation of G1 Cell Cycle Progression: Distinguishing the Restriction Point from a Nutrient-Sensing Cell Growth Checkpoint(s). Genes Cancer 2010, 1, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, J.; Chen, D.; Huang, J.; Feng, B.; Han, S.; Chen, Y.; Song, H.; De, W.; Zhu, Z.; et al. Aurora-A promotes chemoresistance in hepatocelluar carcinoma by targeting NF-kappaB/microRNA-21/PTEN signaling pathway. Oncotarget 2014, 5, 12916–12935. [Google Scholar] [CrossRef]
- Katsha, A.; Belkhiri, A.; Goff, L.; El-Rifai, W. Aurora kinase A in gastrointestinal cancers: Time to target. Mol. Cancer 2015, 14, 106. [Google Scholar] [CrossRef]
- Lu, J.W.; Yang, W.Y.; Tsai, S.M.; Lin, Y.M.; Chang, P.H.; Chen, J.R.; Wang, H.D.; Wu, J.L.; Jin, S.L.; Yuh, C.H. Liver-specific expressions of HBx and src in the p53 mutant trigger hepatocarcinogenesis in zebrafish. PLoS ONE 2013, 8, e76951. [Google Scholar] [CrossRef]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Bankfalvi, A.; Gu, X.; Eberhardt, W.E.; Zeidler, R.; Lang, S.; Brandau, S. Neutrophils Activate Tumoral CORTACTIN to Enhance Progression of Orohypopharynx Carcinoma. Front. Immunol. 2013, 4, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaemmerer, D.; Peter, L.; Lupp, A.; Schulz, S.; Sanger, J.; Baum, R.P.; Prasad, V.; Hommann, M. Comparing of IRS and Her2 as immunohistochemical scoring schemes in gastroenteropancreatic neuroendocrine tumors. Int. J. Clin. Exp. Pathol. 2012, 5, 187–194. [Google Scholar] [PubMed]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/beta-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Waisberg, J.; Saba, G.T. Wnt-/-beta-catenin pathway signaling in human hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2631–2635. [Google Scholar] [CrossRef]
- Taga, M.; Hirooka, E.; Ouchi, T. Essential roles of mTOR/Akt pathway in Aurora-A cell transformation. Int. J. Biol. Sci. 2009, 5, 444–450. [Google Scholar] [CrossRef]
- Lu, J.W.; Hsia, Y.; Yang, W.Y.; Lin, Y.I.; Li, C.C.; Tsai, T.F.; Chang, K.W.; Shieh, G.S.; Tsai, S.F.; Wang, H.D.; et al. Identification of the common regulators for hepatocellular carcinoma induced by hepatitis B virus X antigen in a mouse model. Carcinogenesis 2012, 33, 209–219. [Google Scholar] [CrossRef]
- Lin, H.S.; Huang, Y.L.; Wang, Y.S.; Hsiao, E.; Hsu, T.A.; Shiao, H.Y.; Jiaang, W.T.; Sampurna, B.P.; Lin, K.H.; Wu, M.S.; et al. Identification of Novel Anti-Liver Cancer Small Molecules with Better Therapeutic Index than Sorafenib via Zebrafish Drug Screening Platform. Cancers (Basel) 2019, 11, 739. [Google Scholar] [CrossRef]
- Wrighton, P.J.; Oderberg, I.M.; Goessling, W. There’s Something Fishy About Liver Cancer: Zebrafish Models of Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Horikawa, Y. ChREBP: A glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J. 2008, 55, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carman, G.M.; Han, G.S. Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J. Biol. Chem. 2009, 284, 2593–2597. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.A.; Haas, J.T.; Streeper, R.S.; Stone, S.J.; Kumari, M.; Yang, K.; Han, X.; Brownell, N.; Gross, R.W.; Zechner, R.; et al. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J. Lipid Res. 2011, 52, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavin, R.; Zadra, G.; Loda, M. Metabolic alterations and targeted therapies in prostate cancer. J. Pathol. 2011, 223, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Li, P.; Cao, Y.; Li, Y.; Zhou, L.; Liu, X.; Geng, M. Expression of Wnt-5a and beta-catenin in primary hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3190–3195. [Google Scholar]
- Saiprasad, G.; Chitra, P.; Manikandan, R.; Sudhandiran, G. Hesperidin induces apoptosis and triggers autophagic markers through inhibition of Aurora-A mediated phosphoinositide-3-kinase/Akt/mammalian target of rapamycin and glycogen synthase kinase-3 beta signalling cascades in experimental colon carcinogenesis. Eur. J. Cancer 2014, 50, 2489–2507. [Google Scholar] [CrossRef]
- Kwan, K.M.; Fujimoto, E.; Grabher, C.; Mangum, B.D.; Hardy, M.E.; Campbell, D.S.; Parant, J.M.; Yost, H.J.; Kanki, J.P.; Chien, C.B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007, 236, 3088–3099. [Google Scholar] [CrossRef]
- You, M.S.; Jiang, Y.J.; Yuh, C.H.; Wang, C.M.; Tang, C.H.; Chuang, Y.J.; Lin, B.H.; Wu, J.L.; Hwang, S.P. A Sketch of the Taiwan Zebrafish Core Facility. Zebrafish 2016, 13 (Suppl. 1), S24–S29. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Name | Sequence (5′ to 3′) |
|---|---|---|
| AURKA | Q-AURKA-F | TGGAATATGCACCACTTGGA |
| Q-AURKA-R | ACTGACCACCCAAAATCTGC | |
| actin | Q-actin-F | CTCCATCATGAAGTGCGACGT |
| Q-actin-R | CAGACGGAGTATTTGCGCTCA | |
| pparγ | Q-pparγ- | GGTTTCATTACGGCGTTCAC |
| Q-pparγ-R | TGGTTCACGTCACTGGAGAA | |
| srebp1 | Q-srebp1-F | CATCCACATGGCTCTGAGTG |
| Q-srebp1-R | CTCATCCACAAAGAAGCGGT | |
| chrebp | Q-chrebp-F-2 | GGAGATGGACTCGCTCTTTG |
| Q-chrebp-R-2 | GCAGAGGCTCAAAAGTGTCC | |
| dgat2 | Q-dgat2-F | TGGGGCTTTTTGTAACTTCG |
| Q-dgat2-R | TCTTCCTGGTGCACAGTCC | |
| pap | Q-pap-F | CAGTTCTTCCTGATTGCTGC |
| Q-pap-R | TCCTCAAAGCTTAGTTCGGG | |
| fasn | Q-fasn-F | ATCTGTTCCTGTTCGATGGC |
| Q-fasn-R | AGCATATCTCGGCTGACGTT | |
| col1a1 | Q-col1a1-F | TATTGGTGGTCAGCGTGGTA |
| Q-col1a1-R | TCCTGGAGTACCCTCACGAC | |
| ctgfa | Q-ctgfa-F | TGTGTGTTTGGTGGAATGGT |
| Q-ctgfa-R | GGAGTCACACACCCACTCCT | |
| hpse | Q-hpse-F | GCTCTGGTTTGGAGCTCATC |
| Q-hpse-R | GAAATCCCGACCAAGTTGAA | |
| ccne1 | Q-ccne1-F | TCCCGACACAGGTTACACAA |
| Q-ccne1-R | TTGTCTTTTCCGAGCAGGTT | |
| cdk1 | Q-cdk1-F | CTCTGGGGACCCCTAACAAT |
| Q-cdk1-R | CGGATGTGTCATTGCTTGTC | |
| cdk2 | Q-cdk2-F | CAGCTCTTCCGGATATTTCG |
| Q-cdk2-R | CCGAGATCCTCTTGTTTGGA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.-L.; Su, C.-W.; Huang, Y.-L.; Yang, W.-Y.; Sampurna, B.P.; Ouchi, T.; Lee, K.-L.; Wu, C.-S.; Wang, H.-D.; Yuh, C.-H. A Novel AURKA Mutant-Induced Early-Onset Severe Hepatocarcinogenesis Greater than Wild-Type via Activating Different Pathways in Zebrafish. Cancers 2019, 11, 927. https://doi.org/10.3390/cancers11070927
Su Z-L, Su C-W, Huang Y-L, Yang W-Y, Sampurna BP, Ouchi T, Lee K-L, Wu C-S, Wang H-D, Yuh C-H. A Novel AURKA Mutant-Induced Early-Onset Severe Hepatocarcinogenesis Greater than Wild-Type via Activating Different Pathways in Zebrafish. Cancers. 2019; 11(7):927. https://doi.org/10.3390/cancers11070927
Chicago/Turabian StyleSu, Zhong-Liang, Chien-Wei Su, Yi-Luen Huang, Wan-Yu Yang, Bonifasius Putera Sampurna, Toru Ouchi, Kuan-Lin Lee, Chen-Sheng Wu, Horng-Dar Wang, and Chiou-Hwa Yuh. 2019. "A Novel AURKA Mutant-Induced Early-Onset Severe Hepatocarcinogenesis Greater than Wild-Type via Activating Different Pathways in Zebrafish" Cancers 11, no. 7: 927. https://doi.org/10.3390/cancers11070927
APA StyleSu, Z. -L., Su, C. -W., Huang, Y. -L., Yang, W. -Y., Sampurna, B. P., Ouchi, T., Lee, K. -L., Wu, C. -S., Wang, H. -D., & Yuh, C. -H. (2019). A Novel AURKA Mutant-Induced Early-Onset Severe Hepatocarcinogenesis Greater than Wild-Type via Activating Different Pathways in Zebrafish. Cancers, 11(7), 927. https://doi.org/10.3390/cancers11070927