Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype


Abstract
:1. Introduction
2. Results
2.1. AML Patients Included in the Study
2.2. Lipopolysaccharide Readily Induces Cytokine Expression
2.3. Response towards Agonists Is Linked with Mutations in the Nucleophosmin Gene
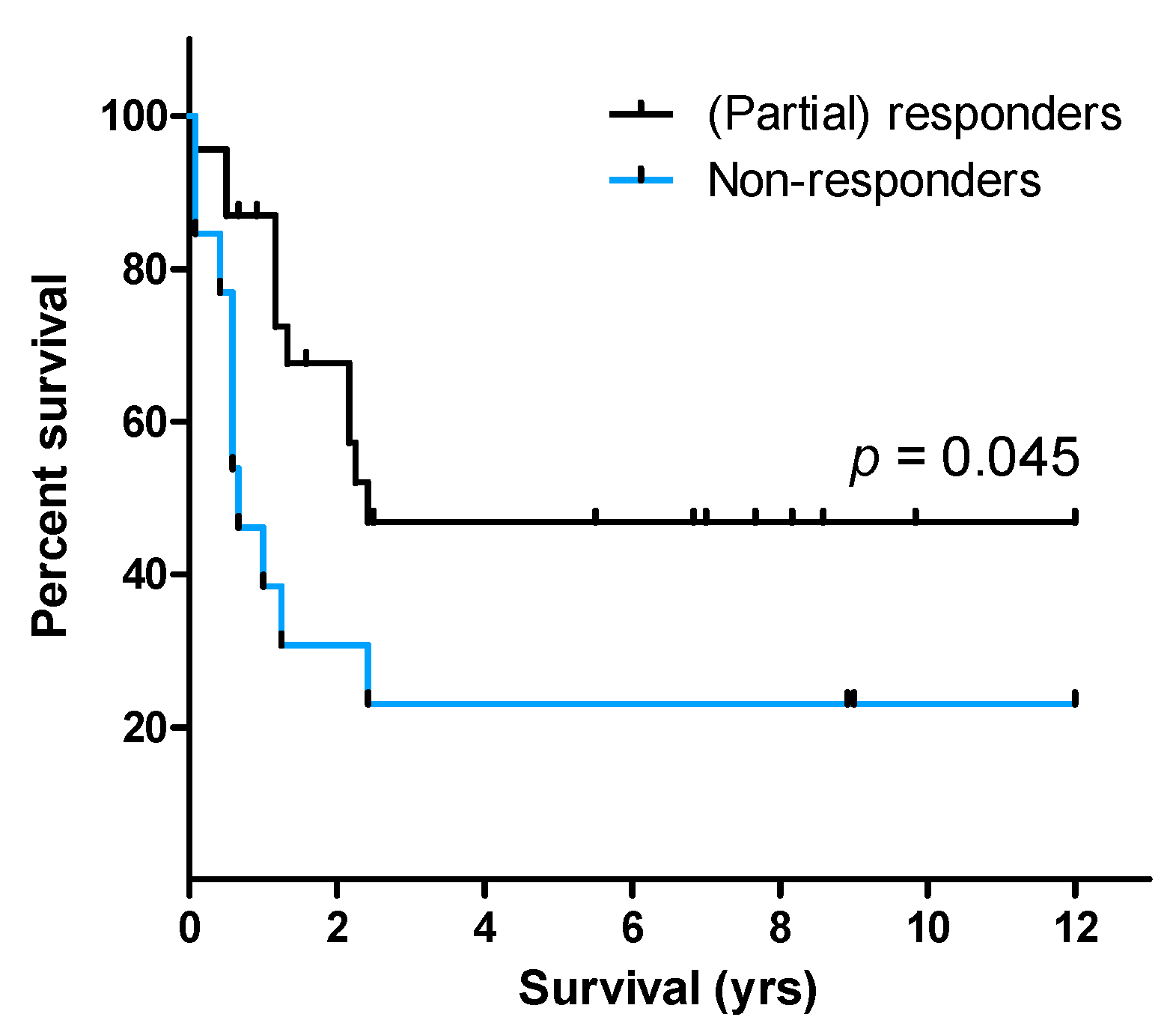
2.4. Response towards the TLR1/2 Agonist Is Linked with Prolonged Patient Survival
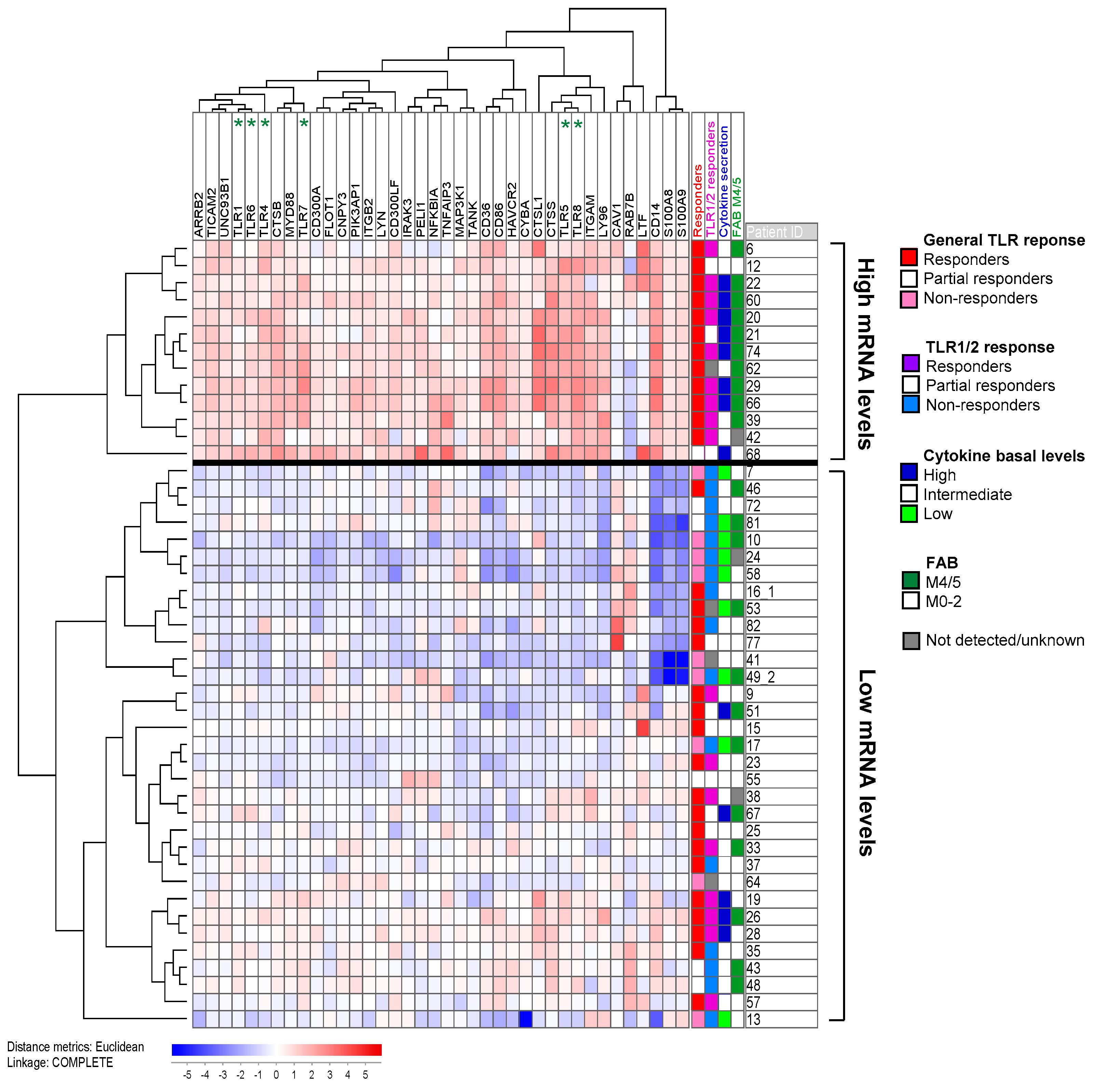
2.5. Response upon TLR Activation Is Correlated with Increased Expression of Genes in the TLR Signaling Pathway and High Constitutive Cytokine Release
2.6. TLR Responders Show Low mRNA Expression of Transcription-Related Proteins
3. Discussion
4. Materials and Methods
4.1. Patients and Cell Preparations
4.2. Reagents
4.3. Analysis of Mediator Levels in Cell Culture Supernatants
4.4. RNA Preparation and Analysis of Global Gene Expression
4.5. Statistical and Bioinformatical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caceres-Cortes, R.J. Blastic leukaemias (AML): A biologist’s view. Cell Biochem. Biophys. 2013, 66, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Mele, L.; Pulsoni, A.; Equitani, F.; Pagano, L. The incidence of secondary leukemias. Haematologica 1999, 84, 937–945. [Google Scholar] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.C.; Choi, S. Recent progress in the development of Toll-like receptor (TLR) antagonists. Expert Opin. Pat. 2016, 26, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Smith, C.; Yin, H. Targeting Toll-like receptors with small molecule agents. Chem. Soc. Rev. 2013, 42, 4859–4866. [Google Scholar] [CrossRef]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Monlish, D.A.; Bhatt, S.T.; Schuettpelz, L.G. The role of toll-like receptors in hematopoietic malignancies. Front. Immunol. 2016, 7, 390. [Google Scholar] [CrossRef] [PubMed]
- Ramzi, M.; Khalafi-Nezhad, A.; Iravani Saadi, M.; Jowkar, Z. Association between TLR2 and TLR4 expression and response to induction therapy in acute myeloid leukemia patients. Int. J. Hematol. Oncol. Stem. Cell Res. 2018, 12, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Rybka, J.; Butrym, A.; Wrobel, T.; Jazwiec, B.; Stefanko, E.; Dobrzynska, O.; Poreba, R.; Kuliczkowski, K. The expression of Toll-like receptors in patients with acute myeloid leukemia treated with induction chemotherapy. Leuk. Res. 2015, 39, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Villamón, E.; González-Fernández, J.; Such, E.; Cervera, J.V.; Gozalbo, D.; Luisa Gil, M. Imiquimod inhibits growth and induces differentiation of myeloid leukemia cell lines. Cancer Cell Int. 2018, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Ignatz-Hoover, J.J.; Wang, H.; Moreton, S.A.; Chakrabarti, A.; Agarwal, M.K.; Sun, K.; Gupta, K.; Wald, D.N. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia 2015, 29, 918–926. [Google Scholar] [CrossRef]
- Eriksson, M.; Peña-Martinez, P.; Ramakrishnan, R.; Chapellier, M.; Högberg, C.; Glowacki, G.; Orsmark-Pietras, C.; Velasco-Hernandez, T.; Lazarevic, V.L.; Juliusson, G.; et al. Agonistic targeting of TLR1/TLR2 induces p38 MAPK-dependent apoptosis and NFkappaB-dependent differentiation of AML cells. Blood Adv. 2017, 1, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hirai, H.; Taniguchi, K.; Shimura, K.; Inaba, T.; Shimazaki, C.; Taniwaki, M.; Imanishi, J. Toll-like receptors (TLRs) are expressed by myeloid leukaemia cell lines, but fail to trigger differentiation in response to the respective TLR ligands. Br. J. Haematol. 2009, 147, 585–587. [Google Scholar] [CrossRef]
- Smits, E.L.; Cools, N.; Lion, E.; Van Camp, K.; Ponsaerts, P.; Berneman, Z.N.; Van Tendeloo, V.F. The Toll-like receptor 7/8 agonist resiquimod greatly increases the immunostimulatory capacity of human acute myeloid leukemia cells. Cancer Immunol. Immunother. 2010, 59, 35–46. [Google Scholar] [CrossRef]
- Smits, E.L.; Ponsaerts, P.; Van de Velde, A.L.; Van Driessche, A.; Cools, N.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Van Tendeloo, V.F. Proinflammatory response of human leukemic cells to dsRNA transfection linked to activation of dendritic cells. Leukemia 2007, 21, 1691–1699. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Øyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Aasebø, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.; Gronningsæter, I.S.; Bartaula-Brevik, S.; Bruserud, Ø. The capacity of long-term in vitro proliferation of acute myeloid leukemia cells supported only by exogenous cytokines is associated with a patient subset with adverse outcome. Cancers 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Na, B.H.; Hoang, T.X.; Kim, J.Y. Hsp90 inhibition reduces TLR5 surface expression and nf-kappab activation in human myeloid leukemia THP-1 cells. Biomed. Res. Int. 2018, 2018, 4319369. [Google Scholar] [CrossRef] [PubMed]
- Bosman, M.C.; Schuringa, J.J.; Vellenga, E. Constitutive NF-kappaB activation in AML: Causes and treatment strategies. Crit. Rev. Oncol. Hematol. 2016, 98, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Breccia, M.; Alimena, G. NF-kappaB as a potential therapeutic target in myelodysplastic syndromes and acute myeloid leukemia. Expert Opin. Targets 2010, 14, 1157–1176. [Google Scholar] [CrossRef]
- Beck, B.; Dorfel, D.; Lichtenegger, F.S.; Geiger, C.; Lindner, L.; Merk, M.; Schendel, D.J.; Subklewe, M. Effects of TLR agonists on maturation and function of 3-day dendritic cells from AML patients in complete remission. J. Transl. Med. 2011, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Nourizadeh, M.; Masoumi, F.; Memarian, A.; Alimoghaddam, K.; Moazzeni, S.M.; Hadjati, J. Synergistic effect of Toll-like receptor 4 and 7/8 agonists is necessary to generate potent blast-derived dendritic cells in Acute Myeloid Leukemia. Leuk. Res. 2012, 36, 1193–1199. [Google Scholar] [CrossRef]
- Nourizadeh, M.; Masoumi, F.; Memarian, A.; Alimoghaddam, K.; Moazzeni, S.M.; Yaghmaie, M.; Hadjati, J. In vitro induction of potent tumor-specific cytotoxic T lymphocytes using TLR agonist-activated AML-DC. Target Oncol. 2014, 9, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Lehner, M.; Bailo, M.; Stachel, D.; Roesler, W.; Parolini, O.; Holter, W. Caspase-8 dependent apoptosis induction in malignant myeloid cells by TLR stimulation in the presence of IFN-alpha. Leuk. Res. 2007, 31, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Li, H.; Messer, K.; Lane, T.A.; Zhou, J.; Ball, E.D. Augmentation of autologous T cell reactivity with acute myeloid leukemia (AML) blasts by Toll-like receptor (TLR) agonists. Cancer Immunol. Immunother. 2015, 64, 737–744. [Google Scholar] [CrossRef]
- Lin, J.; Kato, M.; Nagata, K.; Okuwaki, M. Efficient DNA binding of NF-kappaB requires the chaperone-like function of NPM1. Nucleic Acids Res. 2017, 45, 3707–3723. [Google Scholar] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, Ø.; Wendelbø, Ø.; Paulsen, K. Lipoteichoic acid derived from Enterococcus faecalis modulates the functional characteristics of both normal peripheral blood leukocytes and native human acute myelogenous leukemia blasts. Eur. J. Haematol. 2004, 73, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Tsykunova, G.; Reikvam, H.; Hovland, R.; Bruserud, Ø. The surface molecule signature of primary human acute myeloid leukemia (AML) cells is highly associated with NPM1 mutation status. Leukemia 2012, 26, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Tvedt, T.H.; Nepstad, I.; Rye, K.P.; Hagen, K.M.; Reikvam, H.; Bruserud, Ø. Patients with acute myeloid leukemia can be subclassified based on the constitutive cytokine release of the leukemic cells; the possible clinical relevance and the importance of cellular iron metabolism. Expert Opin. Targets 2017, 21, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Re, F.; Arpinati, M.; Testoni, N.; Ricci, P.; Terragna, C.; Preda, P.; Ruggeri, D.; Senese, B.; Chirumbolo, G.; Martelli, V.; et al. Expression of CD86 in acute myelogenous leukemia is a marker of dendritic/monocytic lineage. Exp. Hematol. 2002, 30, 126–134. [Google Scholar] [CrossRef]
- Laouedj, M.; Tardif, M.R.; Gil, L.; Raquil, M.A.; Lachhab, A.; Pelletier, M.; Tessier, P.A.; Barabe, F. S100A9 induces differentiation of acute myeloid leukemia cells through TLR4. Blood 2017, 129, 1980–1990. [Google Scholar] [CrossRef] [Green Version]
- Honnemyr, M.; Bruserud, Ø.; Brenner, A.K. The constitutive protease release by primary human acute myeloid leukemia cells. J. Cancer Res. Clin. Oncol. 2017, 143, 1985–1998. [Google Scholar] [CrossRef]
- Takamatsu-Ichihara, E.; Kitabayashi, I. The roles of Polycomb group proteins in hematopoietic stem cells and hematological malignancies. Int. J. Hematol. 2016, 103, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwama, A. Polycomb repressive complexes in hematological malignancies. Blood 2017, 130, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudy, N.S.; Fawzy, I.M.; Azmy, E.; Goda, E.F.; Eneen, A.; Abdul Salam, E.M. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood Cells Mol. Dis. 2014, 53, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhang, L.; Li, X.; Chen, F.; Jiang, L.; Yu, G.; Wang, Z.; Yin, C.; Jiang, X.; Zhong, Q.; et al. Higher EZH2 expression is associated with extramedullary infiltration in acute myeloid leukemia. Tumour. Biol. 2016, 37, 11409–11420. [Google Scholar] [CrossRef]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell Mol. Life Sci. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Copeland, R.A.; Keilhack, H. PRC2 and SWI/SNF Chromatin Remodeling Complexes in Health and Disease. Biochemistry 2016, 55, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wan, X.; Zhou, P.; Zhou, X.; Zhang, W.; Hui, X.; Yuan, X.; Ding, X.; Zhu, R.; Meng, G.; et al. The chromatin remodeling subunit Baf200 promotes normal hematopoiesis and inhibits leukemogenesis. J. Hematol. Oncol. 2018, 11, 27. [Google Scholar] [CrossRef]
- Satija, Y.K.; Bhardwaj, A.; Das, S. A portrayal of E3 ubiquitin ligases and deubiquitylases in cancer. Int. J. Cancer 2013, 133, 2759–2768. [Google Scholar] [CrossRef]
- Sahasrabuddhe, A.A.; Elenitoba-Johnson, K.S. Role of the ubiquitin proteasome system in hematologic malignancies. Immunol. Rev. 2015, 263, 224–239. [Google Scholar] [CrossRef]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Brenner, A.K.; Hagen, K.M.; Liseth, K.; Skrede, S.; Hatfield, K.J.; Bruserud, O. The cytokine-mediated crosstalk between primary human acute myeloid cells and mesenchymal stem cells alters the local cytokine network and the global gene expression profile of the mesenchymal cells. Stem. Cell Res. 2015, 15, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Bruserud, Ø.; Ryningen, A.; Wergeland, L.; Glenjen, N.I.; Gjertsen, B.T. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 2004, 89, 391–402. [Google Scholar]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvaer, E.; Hatfield, K.J.; Bruserud, Ø. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Dysvik, B.; Jonassen, I. J-Express: Exploring gene expression data using Java. Bioinformatics 2001, 17, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Salamone, G.V.; Petracca, Y.; Fuxman Bass, J.I.; Rumbo, M.; Nahmod, K.A.; Gabelloni, M.L.; Vermeulen, M.E.; Matteo, M.J.; Geffner, J.R.; Trevani, A.S. Flagellin delays spontaneous human neutrophil apoptosis. Lab. Investig. 2010, 90, 1049–1059. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Adj. HR | 95% CI | p-Value |
|---|---|---|---|
| TLR1/2 (non-responder) 1 | 1.86 | 1.07–3.24 | 0.029 |
| Age (≥60 years) | 1.57 | 0.86–2.88 | 0.145 |
| Etiology (secondary) | 3.05 | 0.62–14.94 | 0.170 |
| Cytogenetics (intermediate/adverse) | 0.56 | 0.14–2.29 | 0.422 |
| Flt3-ITD | 2.53 | 0.80–7.95 | 0.113 |
| NPM1-insertion | 0.81 | 0.21–3.24 | 0.758 |
| Function | Proteins |
|---|---|
| Chaperone, translocation | CNPY3/PRAT4A, UNC93B1 |
| TLR cleavage | CTSB, CTSS |
| Agonists | S100A8, S100A9 |
| Receptors | TLR1, TLR4, TLR5, TLR6, TLR7, TLR8 |
| Co-receptors | CD14, CD36, CD86, LY96/MD2 |
| Adaptors, co-factors | CTSL1, MYD88, PIK3AP1/BCAP, TICAM2/TRAM |
| Downstream targets | CYBA, MAP3K1/MEKK1, PELI1 |
| Positive regulators/feedback pathways | CAV1, FLOT1, LTF |
| Negative regulators/feedback pathways | ARRB2, CD300A, CD300LF, HAVCR2/TIM-3, IRAK3/IRAK-M, ITGAM/CD11b, ITGB2, LYN, NFKBIA, PIK3AP1/BCAP, RAB7B, TANK, TNFAIP3 |
| Protein Task | Protein Group | Protein Names | Protein Names (Putative Task) |
|---|---|---|---|
| Pre-Transcription | |||
| Chromatin remodeling | |||
| Polycomb | ASXL1, BMI1, CBX2, EZH2, SCMH1 | ||
| SWI/SNF | ARID2, CHD8, SMARCC1, SMARCD1 | CHD9 | |
| WD40-repeats | BRWD1, FBXW9, RBBP4, WDR33 | WDR42A | |
| Chromatin modification | SETD1A, ZMYM3 | ||
| Chromatin binding | MED12, MYST3, MYST4, RCOR3 | C14orf106 | |
| Transcription | |||
| TFs | Activation and/or regulation | DACH1, ELF2, FOXJ3, ILF3, MYST4, MZF1, NFX1, RFX7, SCMH1, TP53INP1, ZBTB20, ZNF395, ZNF521, ZNF548, ZNF880 | CCDCC171, KANSL3, RCOR3, UBN2, ZNF521, ZNF669, ZNF700, ZNF763, ZNF789 |
| Transcription repressors | TRIM33, ZNF219, ZNF431, ZNF521 | ||
| Post-Transcription | |||
| Pre-mRNA splicing | SFRS8, SFRS14, SNRNP200, TCF3, WDR33, YTHDC1 | EXOC7, PSIP1 | |
| mRNA degradation | DCP1A | ||
| Ubiquitination | |||
| E3 ubiquitin ligases | CCNB1IP1, DDB1, FBXO46, FBXW9, HECTD2, HECTD4, HERC1, MAGEL2, MARCH5, RNF19A, RNF144, RNF144A, SIAH1 | ||
| Other tasks with ubiquitination | CUL9, KLHL | COMMD3, WDR42A | |
| De-ubiquitination | OTUD5, USP4 | BABAM1 |
| Patient Characteristics, Cell Morphology | Disease Etiology, Cell Morphology | Cell Genetics | |||
|---|---|---|---|---|---|
| Age | De novo AML1 | 55 | Cytogenetics | ||
| Median (yrs) | 66 | Favorable | 7 | ||
| Range (yrs) | 18–92 | Secondary AML | Intermediate | 14 | |
| MDS | 12 | Normal | 38 | ||
| Gender | CMML | 4 | Adverse | 14 | |
| Females | 39 | CML | 1 | n.d. | 10 |
| Males | 44 | CLL | 1 | ||
| MF | 4 | Flt3 mutations3 | |||
| FAB classification | PV | 1 | ITD | 26 | |
| M0 | 5 | Chemotherapy | 1 | Wild-type | 41 |
| M1 | 20 | n.d. | 16 | ||
| M2 | 14 | AML relapse2 | 7 | ||
| M4 | 21 | NPM1 mutations | |||
| M5 | 17 | CD34 receptor | Mutated | 22 | |
| n.d. | 6 | Negative (≤20%) | 25 | Wild-type | 45 |
| Positive (>20%) | 50 | n.d. | 16 | ||
| n.d. | 8 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenner, A.K.; Bruserud, Ø. Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype. Cancers 2019, 11, 973. https://doi.org/10.3390/cancers11070973
Brenner AK, Bruserud Ø. Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype. Cancers. 2019; 11(7):973. https://doi.org/10.3390/cancers11070973
Chicago/Turabian StyleBrenner, Annette K., and Øystein Bruserud. 2019. "Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype" Cancers 11, no. 7: 973. https://doi.org/10.3390/cancers11070973
APA StyleBrenner, A. K., & Bruserud, Ø. (2019). Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype. Cancers, 11(7), 973. https://doi.org/10.3390/cancers11070973