SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis


Abstract
:1. Introduction
2. Epigenetic Regulation Leads to Cellular Differentiation by Chromatin Modification
How DNA Methyltransferases Interact with Histone Modification Proteins to Regulate Gene Expression
3. SETDB1: A Nuclear Transcriptional Regulator
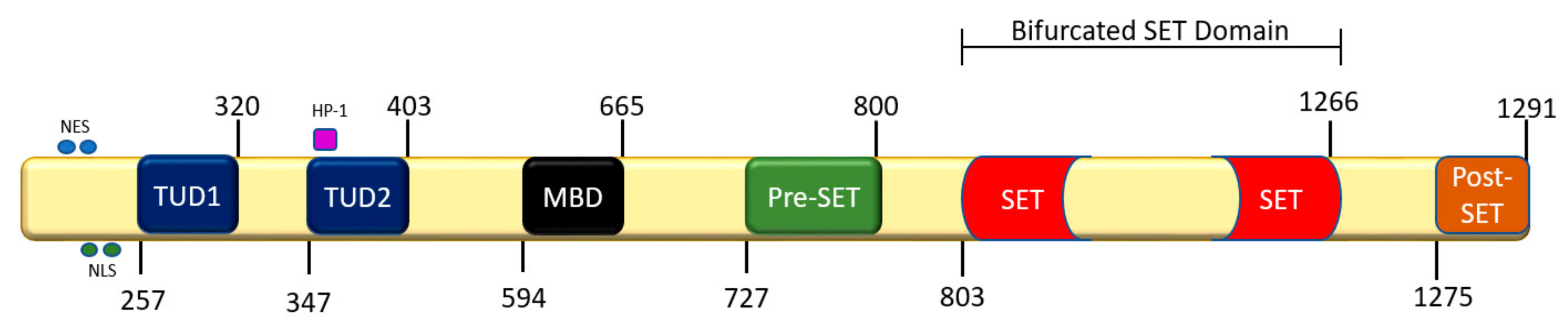
3.1. Structural and Functional Biochemistry of SETDB1
3.2. Localization of SETDB1
3.3. The Nuclear Role of SETDB1
3.3.1. SETDB1 Is Protected against Proteasomal Degradation by ATF71P
3.3.2. Function of SETDB1 Interactions with Promyelocytic Leukemia Nuclear Bodies (PML-NBs)
3.3.3. SETDB1 Has the Ability to Regulate Viral Transcription
4. Epigenomic Functions and Interactions of SETDB1
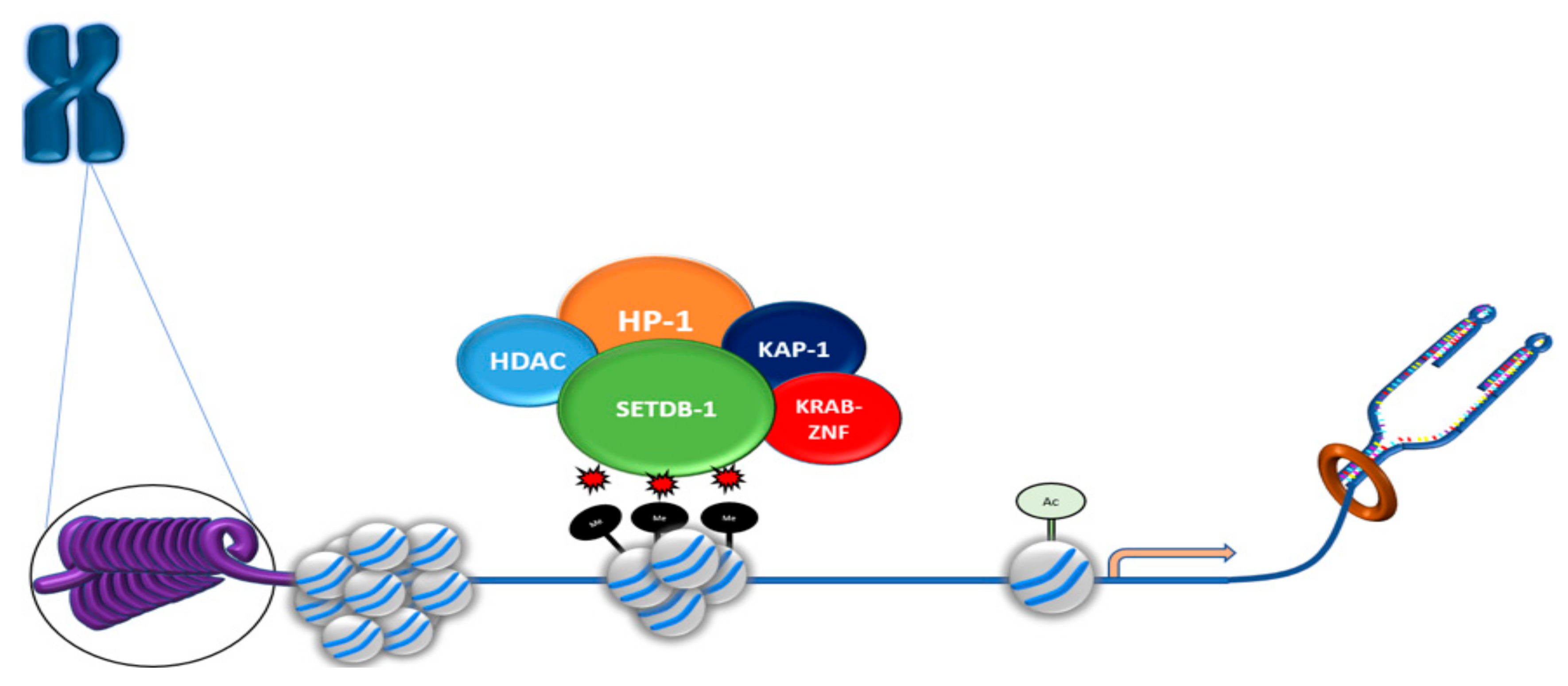
4.1. SETDB1 Is Associated with Transcriptional Modulators
4.2. SETDB1 Interacts with Early Embryological Machinery
4.3. SETDB1 Dictates T-Cell Development and Linage Commitment
5. Structural and Functional Dysregulation of SETDB1 Due to Mutations
6. Epigenetic Influence of SETDB1 in Tumorigenesis
6.1. SETDB1 Is Associated with Cancer Pathway Activation
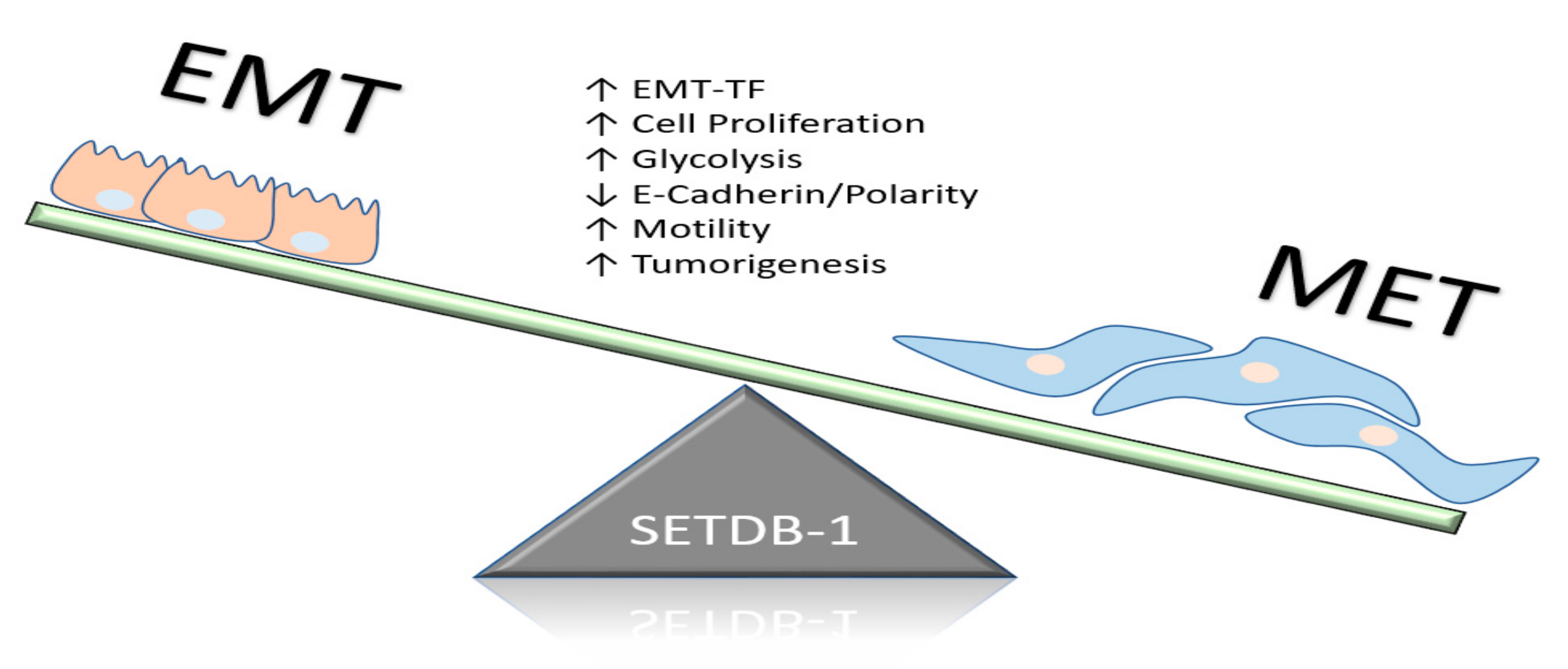
6.2. Epigenetic Influence of SETDB1 in Breast Cancer
6.3. SETDB1 Methyltransferase Activity Sustains Hematopoietic and Progenitor Stem Cell Lineages
6.4. The Influence of SETDB1 on Stem and Cancer Cell Metabolism and Its Consequence on the Warburg Effect
7. SETDB1 as a Therapeutic Target
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Australian Institute of Health and Welfare. Cancer Data in Australia; AIHW: Canberra, Australia, 2019. [Google Scholar]
- Filipova, A.; Seifrtova, M.; Mokry, J.; Dvorak, J.; Rezacova, M.; Filip, S.; Diaz-Garcia, D. Breast cancer and cancer stem cells: a mini-review. Tumori 2014, 100, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev Dyn 2019, 248, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collina, F.; Di Bonito, M.; Li Bergolis, V.; De Laurentiis, M.; Vitagliano, C.; Cerrone, M.; Nuzzo, F.; Cantile, M.; Botti, G. Prognostic Value of Cancer Stem Cells Markers in Triple-Negative Breast Cancer. Biomed Res Int. 2015, 2015, 158682. [Google Scholar] [CrossRef]
- Lee, W.J.; Kim, S.C.; Yoon, J.H.; Yoon, S.J.; Lim, J.; Kim, Y.S.; Kwon, S.W.; Park, J.H. Meta-Analysis of Tumor Stem-Like Breast Cancer Cells Using Gene Set and Network Analysis. PLoS ONE 2016, 11, e0148818. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Lin, C.Y.; Barry-Holson, K.Q.; Allison, K.H. Breast cancer stem cells: are we ready to go from bench to bedside? Histopathology 2016, 68, 119–137. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Torres, I.O.; Fujimori, D.G. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr. Opin. Struct. Biol. 2015, 35, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntean, A.G.; Hess, J.L. Epigenetic dysregulation in cancer. Am. J. Pathol. 2009, 175, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.K.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xia, L.; Wu, D.Y.; Wang, H.; Chansky, H.A.; Schubach, W.H.; Hickstein, D.D.; Zhang, Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 2002, 21, 148. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Kim, K.; Kim, S.-K.; Oh, J.-H.; Min, J.-K.; Jung, C.-R.; Son, M.-Y.; Kim, D.-S.; Cho, H.-S. SETDB1 regulates SMAD7 expression for breast cancer metastasis. BMB Rep. 2019, 52, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Suriyamurthy, S.; Baker, D.; Ten Dijke, P.; Iyengar, P.V. Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer. Cancers 2019, 11, 726. [Google Scholar] [CrossRef]
- Wu, F.; McCuaig, D.R.; Sutton, R.C.; Tan, H.A.; Jeelall, Y.; Bean, G.E.; Dai, J.; Prasanna, T.; Batham, J.; Malik, L.; et al. Nuclear-Biased DUSP6 Expression is Associated with Cancer Spreading Including Brain Metastasis in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Wozniak, G.G.; Strahl, B.D. Hitting the ‘mark’: Interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2014, 1839, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rauch, T.; Chen, Z.-X.; Szabó, P.E.; Riggs, A.D.; Pfeifer, G.P. The Histone Methyltransferase SETDB1 and the DNA Methyltransferase DNMT3A Interact Directly and Localize to Promoters Silenced in Cancer Cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniskan, H.Ü.; Konze, K.D.; Jin, J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, F.; Ding, J.; Liang, Y.; Zhan, Z.; Zhan, Y.; Chen, L.-H.; Ding, Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J. Cancer 2017, 8, 3318–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Gonzalez-Hurtado, E.; Martinez, E. Core promoter-specific gene regulation: TATA box selectivity and Initiator-dependent bi-directionality of serum response factor-activated transcription. Biochim. Biophys. Acta 2016, 1859, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Romier, C.; Cocchiarella, F.; Mantovani, R.; Moras, D. The NF-YB/NF-YC Structure Gives Insight into DNA Binding and Transcription Regulation by CCAAT Factor NF-Y. J. Biol. Chem. 2003, 278, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, M.L.; Chansky, H.A.; Zielinska-Kwiatkowska, A.; Matsui, Y.; Yang, L. Genomic structure and expression of the mouse ESET gene encoding an ERG-associated histone methyltransferase with a SET domain. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2003, 1629, 8–14. [Google Scholar] [CrossRef]
- Cho, S.; Park, J.S.; Kang, Y.-K. Regulated nuclear entry of over-expressed Setdb1. Genes Cells 2013, 18, 694–703. [Google Scholar] [CrossRef]
- Yeap, L.-S.; Hayashi, K.; Surani, M.A. ERG-associated protein with SET domain (ESET)-Oct4 interaction regulates pluripotency and represses the trophectoderm lineage. Epigenetics Chromatin 2009, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Loyola, A.; Tagami, H.; Bonaldi, T.; Roche, D.; Quivy, J.P.; Imhof, A.; Nakatani, Y.; Dent, S.Y.R.; Almouzni, G. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009, 10, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-K. SETDB1 in Early Embryos and Embryonic Stem Cells. Curr. Issues Mol. Biol. 2014, 17, 1–10. [Google Scholar] [PubMed]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)—A novel class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Park, J.S.; Kang, Y.-K. Dual functions of histone-lysine N-methyltransferase Setdb1 protein at promyelocytic leukemia-nuclear body (PML-NB): maintaining PML-NB structure and regulating the expression of its associated genes. J. Biol. Chem. 2011, 286, 41115–41124. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Watanabe, S.; Ichimura, T.; Ohkuma, Y.; Chiba, T.; Saya, H.; Nakao, M. MCAF Mediates MBD1-Dependent Transcriptional Repression. Mol. Cell. Biol. 2003, 23, 2834. [Google Scholar] [CrossRef] [PubMed]
- Basavapathruni, A.; Gureasko, J.; Porter Scott, M.; Hermans, W.; Godbole, A.; Leland, P.A.; Boriack-Sjodin, P.A.; Wigle, T.J.; Copeland, R.A.; Riera, T.V. Characterization of the Enzymatic Activity of SETDB1 and Its 1:1 Complex with ATF7IP. Biochemistry 2016, 55, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Timms, R.T.; Tchasovnikarova, I.A.; Antrobus, R.; Dougan, G.; Lehner, P.J. ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex. Cell Rep. 2016, 17, 653–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Mei, Q.; Zielinska-Kwiatkowska, A.; Matsui, Y.; Blackburn, M.L.; Benedetti, D.; Krumm, A.A.; Taborsky, G.J., Jr.; Chansky, H.A. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem. J. 2003, 369, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Dellaire, G.; Bazett-Jones, D.P. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. BioEssays 2004, 26, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Bohan, C.A.; Kashanchi, F.; Ensoli, B.; Buonaguro, L.; Boris-Lawrie, K.A.; Brady, J.N. Analysis of Tat transactivation of human immunodeficiency virus transcription in vitro. Gene Expr. 2018, 2, 391–407. [Google Scholar]
- Poletti, V.; Mavilio, F. Interactions between Retroviruses and the Host Cell Genome. Mol. Ther. Methods Clin. Dev. 2017, 8, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanth, A.V.; Maniswami, R.R.; Prashanth, S.; Govindaraj, H.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. Emerging role of SETDB1 as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Keniry, A.; Gearing, L.J.; Jansz, N.; Liu, J.; Holik, A.Z.; Hickey, P.F.; Kinkel, S.A.; Moore, D.L.; Breslin, K.; Chen, K.; et al. Setdb1-mediated H3K9 methylation is enriched on the inactive X and plays a role in its epigenetic silencing. Epigenetics Chromatin 2016, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.-M.; Garruss, A.; Shilatifard, A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J., 3rd. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Krüppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Cell. Biol. 1999, 19, 4366–4378. [Google Scholar]
- Wiznerowicz, M.; Jakobsson, J.; Szulc, J.; Liao, S.; Quazzola, A.; Beermann, F.; Aebischer, P.; Trono, D. The Krüppel-associated Box Repressor Domain Can Trigger de Novo Promoter Methylation during Mouse Early Embryogenesis. J. Biol. Chem. 2007, 282, 34535–34541. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, H.M.; Kapopoulou, A.; Corsinotti, A.; Fasching, L.; Macfarlan, T.S.; Tarabay, Y.; Viville, S.; Jakobsson, J.; Pfaff, S.L.; Trono, D. TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res. 2013, 23, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.E.; Kang, Y.-K.; Beppu, H.; Lei, H.; Li, E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 2004, 24, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eymery, A.; Liu, Z.; Ozonov, E.A.; Stadler, M.B.; Peters, A.H.F.M. The methyltransferase Setdb1 is essential for meiosis and mitosis in mouse oocytes and early embryos. Development 2016, 143, 2767–2779. [Google Scholar] [CrossRef] [PubMed]
- Adoue, V.; Binet, B.; Malbec, A.; Fourquet, J.; Romagnoli, P.; van Meerwijk, J.P.M.; Amigorena, S.; Joffre, O.P. The Histone Methyltransferase SETDB1 Controls T Helper Cell Lineage Integrity by Repressing Endogenous Retroviruses. Immunity 2019, 50, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Takikita, S.; Muro, R.; Takai, T.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Oda, H.; Kitajima, M.; Oshima, K.; Hattori, M.; et al. A Histone Methyltransferase ESET Is Critical for T Cell Development. J. Immunol. 2016, 197, 2269. [Google Scholar] [CrossRef] [PubMed]
- Melssen, M.; Slingluff, C.L. Vaccines targeting helper T cells for cancer immunotherapy. Curr. Opin. Immunol. 2017, 47, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-Y.; Ding, L.-W.; Xiao, J.-F.; Chien, W.; Lim, S.-L.; Hattori, N.; Goodglick, L.; Chia, D.; Mah, V.; Alavi, M.; et al. SETDB1 accelerates tumourigenesis by regulating the WNT signalling pathway. J. Pathol. 2015, 235, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.-H.; Jen, K.-Y.; Fang, L.T.; Jones, K.; et al. Whole exome and targeted deep sequencing identify genome-wide allelic loss and frequent SETDB1 mutations in malignant pleural mesotheliomas. Oncotarget 2016, 7, 8321–8331. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu-Monette, Z.Y.; Jabbar, K.J.; Shen, Q.; Manyam, G.C.; Tzankov, A.; Visco, C.; Wang, J.; Montes-Moreno, S.; Dybkær, K.; et al. AKT Hyperactivation and the Potential of AKT-Targeted Therapy in Diffuse Large B-Cell Lymphoma. Am. J. Pathol. 2017, 187, 1700–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Long, J.; Gao, Y.; Zhang, W.; Han, F.; Xu, C.; Sun, L.; Yang, S.-C.; Lan, J.; Hou, Z.; et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019, 21, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Dai, X.; Laurent, B.; Zheng, N.; Gan, W.; Zhang, J.; Guo, A.; Yuan, M.; Liu, P.; Asara, J.M.; et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019, 21, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, Inhibited Indirectly by LincRNA HOTAIR, Directly Inhibits SETDB1 and Reverses the EMT of Breast Cancer Stem Cells by Downregulating the STAT3 Pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Martinez de Paz, A.; Simó-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vázquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Fan, B.; Guo, Q.; Li, Y.; Chen, R.; Lv, N.; Diao, Y.; Luo, Y. Knockdown of SETDB1 inhibits breast cancer progression by miR-381-3p-related regulation. Bone Res. 2018, 51, 39. [Google Scholar] [CrossRef]
- Du, D.; Katsuno, Y.; Meyer, D.; Budi, E.H.; Chen, S.-H.; Koeppen, H.; Wang, H.; Akhurst, R.J.; Derynck, R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef]
- Iglesias, J.M.; Gumuzio, J.; Martin, A.G. Linking Pluripotency Reprogramming and Cancer. Stem Cells Transl. Med. 2017, 6, 335–339. [Google Scholar] [CrossRef]
- Miyagi, S.; Koide, S.; Saraya, A.; Wendt, G.R.; Oshima, M.; Konuma, T.; Yamazaki, S.; Mochizuki-Kashio, M.; Nakajima-Takagi, Y.; Wang, C.; et al. The TIF1β-HP1 system maintains transcriptional integrity of hematopoietic stem cells. Stem Cell Rep. 2014, 2, 145–152. [Google Scholar] [CrossRef]
- Ugarte, F.; Sousae, R.; Cinquin, B.; Martin, E.W.; Krietsch, J.; Sanchez, G.; Inman, M.; Tsang, H.; Warr, M.; Passegué, E.; et al. Progressive Chromatin Condensation and H3K9 Methylation Regulate the Differentiation of Embryonic and Hematopoietic Stem Cells. Stem Cell Rep. 2015, 5, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, S.; Pathak, S.S.; Maitra, S.; Khandelwal, N.; Chandra, K.B.; Kumar, A. Chapter Four - Epigenetic Regulatory Mechanisms in Stress-Induced Behavior. In International Review of Neurobiology; Pandey, S.C., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 115, pp. 117–154. [Google Scholar]
- Lehnertz, B.; Pabst, C.; Su, L.; Miller, M.; Liu, F.; Yi, L.; Zhang, R.; Krosl, J.; Yung, E.; Kirschner, J.; et al. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 2014, 28, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, S.; Oshima, M.; Takubo, K.; Yamazaki, S.; Nitta, E.; Saraya, A.; Aoyama, K.; Kato, Y.; Miyagi, S.; Nakajima-Takagi, Y.; et al. Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood 2016, 128, 638. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen Physiol 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of Drug Resistance in Cancer Chemotherapy. Med Princ. Pract. 2005, 14 (Suppl. 1), 35–48. [Google Scholar] [CrossRef]
- Yang, M.-H.; Imrali, A.; Heeschen, C. Circulating cancer stem cells: the importance to select. Chin. J. Cancer Res. 2015, 27, 437–449. [Google Scholar] [CrossRef]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef]
- Lee, J.-K.; Kim, K.-C. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.-J.; Kim, K.-A.; Kim, K.-C. p53 Down-regulates SETDB1 gene expression during paclitaxel induced-cell death. Biochem. Biophys. Res. Commun. 2014, 446, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.; Abdullah, L.N.; Pang, Q.Y.; Jha, S.; Chow, E.K.-H.; Yang, H.; Kato, H.; Poellinger, L.; Ueda, J.; Lee, K.L. Inhibition of the H3K9 methyltransferase G9A attenuates oncogenicity and activates the hypoxia signaling pathway. PLoS ONE 2017, 12, e0188051. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Giaccia, A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Rao, R.; Balusu, R.; Ganguly, S.; Tao, J.; Sotomayor, E.; Mudunuru, U.; Smith, J.E.; Hembruff, S.L.; Atadja, P.; et al. Superior Efficacy of a Combined Epigenetic Therapy against Human Mantle Cell Lymphoma Cells. Cancer Res. 2012, 18, 6227–6238. [Google Scholar] [CrossRef]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Suzuki, E.; Negishi, M.; Saraya, A.; Miyagi, S.; Konuma, T.; Tanaka, S.; Tada, M.; Kanai, F.; Imazeki, F.; et al. 3-Deazaneplanocin A is a promising therapeutic agent for the eradication of tumor-initiating hepatocellular carcinoma cells. Int. J. Cancer 2012, 130, 2557–2567. [Google Scholar] [CrossRef]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Quinn, M.; Plebanski, M. Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer. Biomed Res. Int. 2015, 2015, 413076. [Google Scholar] [CrossRef]
- Safe, S.; Abbruzzese, J.; Abdelrahim, M.; Hedrick, E. Specificity Protein Transcription Factors and Cancer: Opportunities for Drug Development. Cancer Prev. Res. 2018, 11, 371. [Google Scholar] [CrossRef]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. BCR 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, E.L.; Bunting, K.L.; He, Y.Q.; Li, J.; Phetsouphanh, C.; Seddiki, N.; Zafar, A.; Hindmarsh, E.J.; Parish, C.R.; Kelleher, A.D.; et al. Chromatin-associated protein kinase C-θ regulates an inducible gene expression program and microRNAs in human T lymphocytes. Mol. Cell 2011, 41, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wu, F.; Hardy, K.; Li, J.; Tu, W.J.; McCuaig, R.; Harris, J.; Khanna, K.K.; Attema, J.; Gregory, P.A.; et al. Chromatinized protein kinase C-θ directly regulates inducible genes in epithelial to mesenchymal transition and breast cancer stem cells. Mol. Cell Biol. 2014, 34, 2961–2980. [Google Scholar] [CrossRef] [PubMed]
- Boulding, T.; Wu, F.; McCuaig, R.; Dunn, J.; Sutton, C.R.; Hardy, K.; Tu, W.; Bullman, A.; Yip, D.; Dahlstrom, J.E.; et al. Differential Roles for DUSP Family Members in Epithelial-to-Mesenchymal Transition and Cancer Stem Cell Regulation in Breast Cancer. PLoS ONE 2016, 11, e0148065. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Location | Nucleotide Change AND Mutation Type | Effect |
|---|---|---|---|
| H1224K | C-terminal (SET domain) | Impaired Histone H3 (H3)-methylase activity Accelerated melanoma | |
| C1226A | C-terminal (SET domain) | Impaired H3-methylase activity Accelerated melanoma | |
| C1279Y | C-terminal (SET domain) | Impaired H3-methylase activity | |
| Y249X | N-terminal | 747 T > A Nonsense | Loss of function Multiple primary melanoma (MPM) development |
| V132FS | N-terminal | 395–399del5 Frameshift | Loss of function Premature stop codon indicated in MPM development |
| G869E | C-terminal (Bifurcated SET) | 2606 G > A Missense | |
| C911F | C-terminal (Bifurcated SET) | 2732 G > T Missense | Unknown |
| S947C | C-terminal (Bifurcated SET) | 2840 C > G Missense | Unknown |
| P226RFSX4 P226RFSX4 | N-terminal N-terminal | 677–693del17 Frameshift (Duplicate) | Loss of function Duplicate mutation |
| F1250DEL | C-terminal (Post-SET) | 3747–3749del In-frame deletion | Unknown |
| K674SFSX73 | C-terminal (Pre-SET-MBD aa sequence) | 2020del A Frameshift | Loss of function |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers 2019, 11, 1143. https://doi.org/10.3390/cancers11081143
Batham J, Lim PS, Rao S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers. 2019; 11(8):1143. https://doi.org/10.3390/cancers11081143
Chicago/Turabian StyleBatham, Jacob, Pek Siew Lim, and Sudha Rao. 2019. "SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis" Cancers 11, no. 8: 1143. https://doi.org/10.3390/cancers11081143
APA StyleBatham, J., Lim, P. S., & Rao, S. (2019). SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers, 11(8), 1143. https://doi.org/10.3390/cancers11081143