The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer



Abstract
:1. Introduction
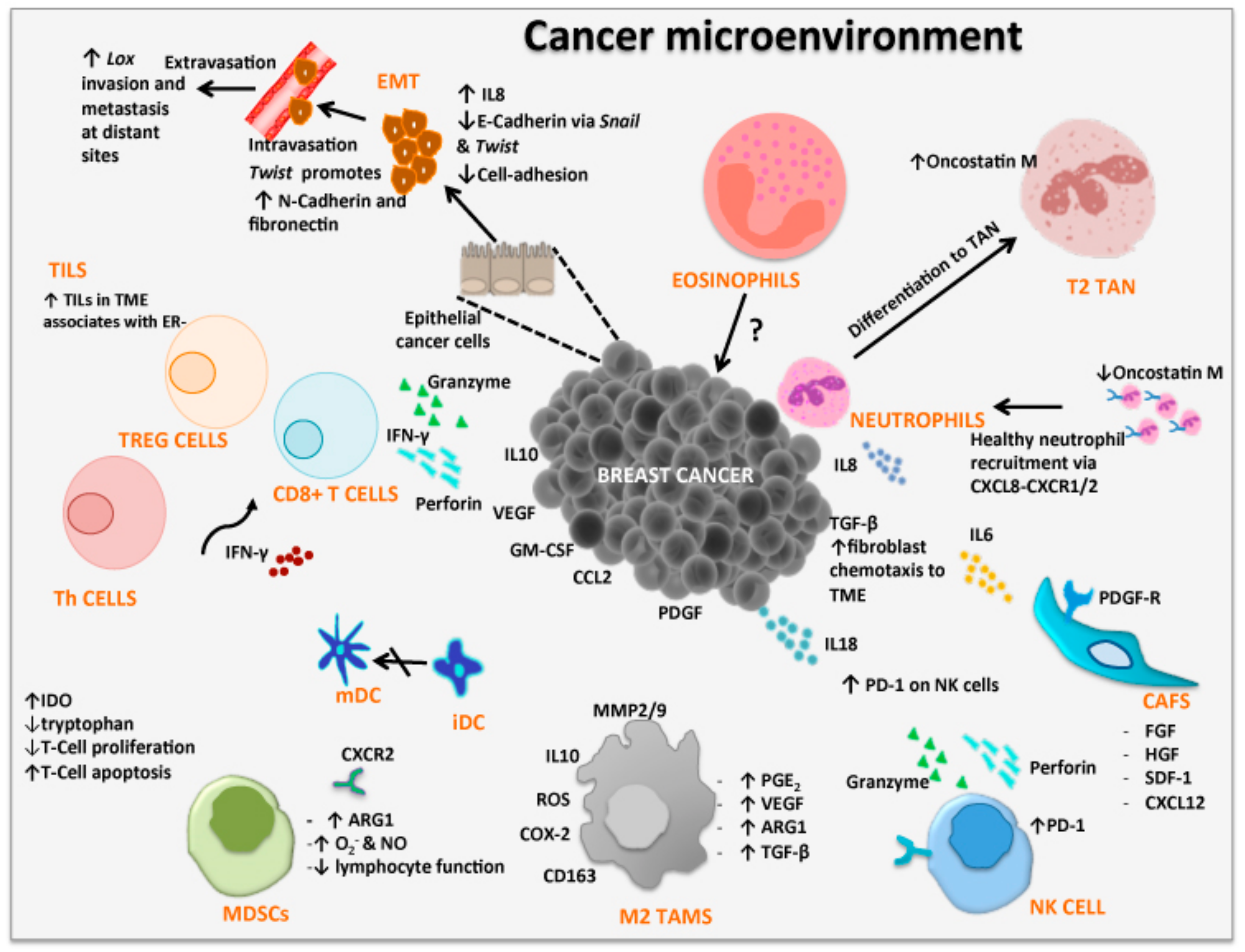
2. Tumor Microenvironment
2.1. Epithelial-Mesenchymal Transition
2.2. Tumor-Infiltrating Lymphocytes
2.3. Cancer-Associated Fibroblasts
2.4. Tumor-Associated Macrophages
2.5. Tumor-Associated Neutrophils
2.6. Tumor-Associated Eosinophils
2.7. Myeloid-Derived Suppressor Cells
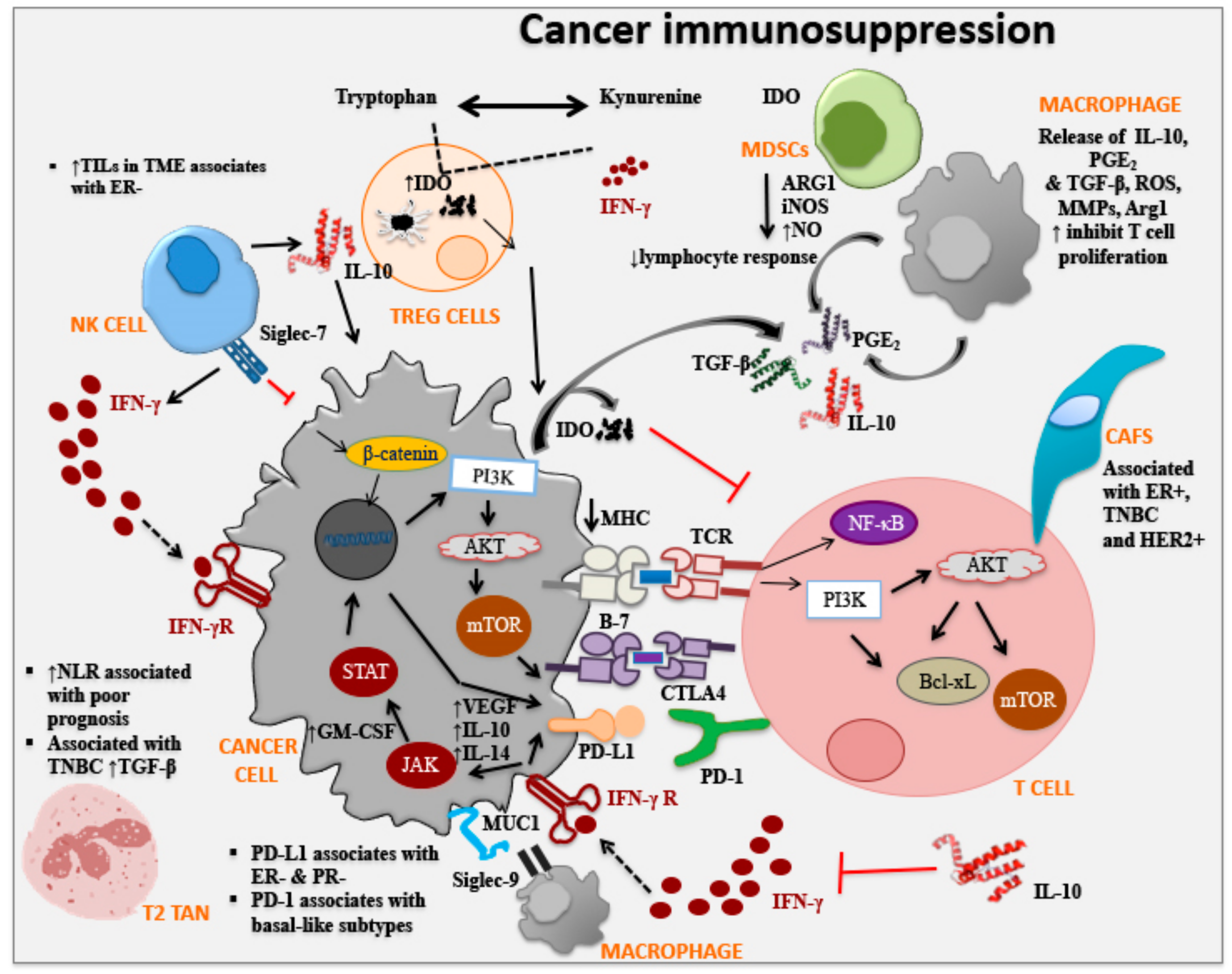
3. Immune Checkpoint Molecules and Breast Cancer
3.1. Cytotoxic T Lymphocyte-Associated Protein 4
3.2. Programmed Cell Death Protein 1
3.3. Indoleamine 2,3-Dioxygenase
3.4. Sialic Acid-Binding Immunoglobulin-Type Lectin
4. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boyle, P.; Ferlay, J. Mortality and survival in breast and colorectal cancer. Nat. Clin. Pract. Oncol. 2005, 2, 424. [Google Scholar] [CrossRef] [PubMed]
- Pudkasam, S.; Tangalakis, K.; Chinlumprasert, N.; Apostolopoulos, V.; Stojanovska, L. Breast cancer and exercise: The role of adiposity and immune markers. Maturitas 2017, 105, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.A.; Duffy, S.W.; Gabe, R.; Tabar, L.; Yen, A.M.; Chen, T.H. The randomized trials of breast cancer screening: What have we learned? Radiol. Clin. North Am. 2004, 42, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Smith, G.L.; Hurria, A.; Hortobagyi, G.N.; Buchholz, T.A. Future of cancer incidence in the United States: Burdens upon an aging, changing nation. J. Clin. Oncol. 2009, 27, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Burness, M.L.; Van Poznak, C. Metastatic breast cancer: Clinical considerations. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 963–970. [Google Scholar]
- Lal, S.; McCart Reed, A.E.; de Luca, X.M.; Simpson, P.T. Molecular signatures in breast cancer. Methods 2017, 131, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakha, E.A.; Reis-Filho, J.S.; Baehner, F.; Dabbs, D.J.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Breast cancer prognostic classification in the molecular era: The role of histological grade. Breast Cancer Res. 2010, 12, 207. [Google Scholar] [CrossRef]
- Weigelt, B.; Reis-Filho, J.S. Histological and molecular types of breast cancer: Is there a unifying taxonomy? Nat. Rev. Clin. Oncol. 2009, 6, 718–730. [Google Scholar] [CrossRef]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2010, 220, 263–280. [Google Scholar] [CrossRef]
- Geyer, F.C.; Reis-Filho, J.S. Microarray-based gene expression profiling as a clinical tool for breast cancer management: Are we there yet? Int. J. Surg. Pathol. 2009, 17, 285–302. [Google Scholar] [CrossRef]
- Geyer, F.C.; Lopez-Garcia, M.A.; Lambros, M.B.; Reis-Filho, J.S. Genetic characterization of breast cancer and implications for clinical management. J. Cell. Mol. Med. 2009, 13, 4090–4103. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the Nervous System in Tumor Angiogenesis. Cancer Microenviron. 2018, 11, 1–11. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 5. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Crosstalk between cancer and the neuro-immune system. J. Neuroimmunol. 2018, 315, 15–23. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Nurgali, K.; Apostolopoulos, V. The mechanisms tumor cells utilize to evade the host’s immune system. Maturitas 2017, 105, 8–15. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Nurgali, K.; Apostolopoulos, V. PD-1/PD-L1 in disease. Immunotherapy 2018, 10, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Hudak, J.E.; Canham, S.M.; Bertozzi, C.R. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat. Chem. Biol. 2014, 10, 69–75. [Google Scholar] [CrossRef]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.D.; Klausing, S.; Hillier, M.; Maher, J.; Noll, T.; Crocker, P.R.; et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulos, V.; Stojanovska, L.; Gargosky, S.E. MUC1 (CD227): A multi-tasked molecule. Cell. Mol. Life Sci. CMLS 2015, 72, 4475–4500. [Google Scholar] [CrossRef]
- Apostolopoulos, V. Peptide-based vaccines for cancer: Are we choosing the right peptides? Expert Rev. Vaccines 2009, 8, 259–260. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; McKenzie, I.F. Cellular mucins: Targets for immunotherapy. Crit. Rev. Immunol. 1994, 14, 293–309. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; McKenzie, I.F.; Pietersz, G.A. Breast cancer immunotherapy: Current status and future prospects. Immunol. Cell Biol. 1996, 74, 457–464. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; McKenzie, I.F.C. Cellular Mucins: Targets for Immunotherapy. Crit. Rev. Immunol. 2017, 37, 421–437. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; McKenzie, I.F. MUC1 and breast cancer. Curr. Opin. Mol. Ther. 1999, 1, 98–103. [Google Scholar]
- Apostolopoulos, V.; Osinski, C.; McKenzie, I.F. MUC1 cross-reactive Galα(1,3)Gal antibodies in humans switch immune responses from cellular to humoral. Nat. Med. 1998, 4, 315–320. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Gordon, S.; Martinez-Pomares, L.; McKenzie, I.F. Aldehyde-mannan antigen complexes target the MHC class I antigen-presentation pathway. Eur. J. Immunol. 2000, 30, 1714–1723. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Loveland, B.E.; Sandrin, M.S.; McKenzie, I.F. Oxidative/reductive conjugation of mannan to antigen selects for T1 or T2 immune responses. Proc. Natl. Acad. Sci. USA 1995, 92, 10128–10132. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; McKenzie, I.F. Cell-mediated immune responses to MUC1 fusion protein coupled to mannan. Vaccine 1996, 14, 930–938. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Sandrin, M.S.; McKenzie, I.F. Mimics and cross reactions of relevance to tumour immunotherapy. Vaccine 1999, 18, 268–275. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Sandrin, M.S.; McKenzie, I.F. Carbohydrate/peptide mimics: Effect on MUC1 cancer immunotherapy. J. Mol. Med. 1999, 77, 427–436. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Xing, P.X.; McKenzie, I.F. Murine immune response to cells transfected with human MUC1: Immunization with cellular and synthetic antigens. Cancer Res. 1994, 54, 5186–5193. [Google Scholar]
- Apostolopoulos, V.; Yu, M.; Corper, A.L.; Teyton, L.; Pietersz, G.A.; McKenzie, I.F.; Wilson, I.A.; Plebanski, M. Crystal structure of a non-canonical low-affinity peptide complexed with MHC class I: A new approach for vaccine design. J. Mol. Biol. 2002, 318, 1293–1305. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Yuriev, E.; Ramsland, P.A.; Halton, J.; Osinski, C.; Li, W.; Plebanski, M.; Paulsen, H.; McKenzie, I.F. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc. Natl. Acad. Sci. USA 2003, 100, 15029–15034. [Google Scholar] [CrossRef] [Green Version]
- Lazoura, E.; Apostolopoulos, V. Rational Peptide-based vaccine design for cancer immunotherapeutic applications. Curr. Med. Chem. 2005, 12, 629–639. [Google Scholar] [CrossRef]
- Lees, C.J.; Apostolopoulos, V.; Acres, B.; Ong, C.S.; Popovski, V.; McKenzie, I.F. The effect of T1 and T2 cytokines on the cytotoxic T cell response to mannan-MUC1. Cancer Immunol. Immunother. 2000, 48, 644–652. [Google Scholar] [CrossRef]
- Lees, C.J.; Apostolopoulos, V.; Acres, B.; Ramshaw, I.; Ramsay, A.; Ong, C.S.; McKenzie, I.F. Immunotherapy with mannan-MUC1 and IL-12 in MUC1 transgenic mice. Vaccine 2000, 19, 158–162. [Google Scholar] [CrossRef]
- Lees, C.J.; Apostolopoulos, V.; McKenzie, I.F. Cytokine production from murine CD4 and CD8 cells after mannan-MUC1 immunization. J. Interferon Cytokine Res. 1999, 19, 1373–1379. [Google Scholar] [CrossRef]
- Lees, C.J.; Smorodinsky, N.; Horn, G.; Wreschner, D.H.; McKenzie, I.F.; Pietersz, G.; Stojanovska, L.; Apostolopoulos, V. MUC1 immunotherapy against a metastatic mammary adenocarcinoma model: Importance of IFN-gamma. Prilozi 2016, 37, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Sheng, K.C.; Kalkanidis, M.; Pouniotis, D.S.; Esparon, S.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G.A. Delivery of antigen using a novel mannosylated dendrimer potentiates immunogenicity in vitro and in vivo. Eur. J. Immunol. 2008, 38, 424–436. [Google Scholar] [CrossRef]
- Son, H.Y.; Apostolopoulos, V.; Chung, J.K.; Kim, C.W.; Park, J.U. Protective efficacy of a plasmid DNA vaccine against transgene-specific tumors by Th1 cellular immune responses after intradermal injection. Cell. Immunol. 2018, 329, 17–26. [Google Scholar] [CrossRef]
- Tang, C.K.; Apostolopoulos, V. Strategies used for MUC1 immunotherapy: Preclinical studies. Expert Rev. Vaccines 2008, 7, 951–962. [Google Scholar] [CrossRef]
- Tang, C.K.; Sheng, K.C.; Pouniotis, D.; Esparon, S.; Son, H.Y.; Kim, C.W.; Pietersz, G.A.; Apostolopoulos, V. Oxidized and reduced mannan mediated MUC1 DNA immunization induce effective anti-tumor responses. Vaccine 2008, 26, 3827–3834. [Google Scholar] [CrossRef]
- Karanikas, V.; Hwang, L.A.; Pearson, J.; Ong, C.S.; Apostolopoulos, V.; Vaughan, H.; Xing, P.X.; Jamieson, G.; Pietersz, G.; Tait, B.; et al. Antibody and T cell responses of patients with adenocarcinoma immunized with mannan-MUC1 fusion protein. J. Clin. Investig. 1997, 100, 2783–2792. [Google Scholar] [CrossRef]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.X.; Pietersz, G.A.; Apostolopoulos, V.; Vaughan, H.; Karanikas, V.; et al. Mannan-MUC1-pulsed dendritic cell immunotherapy: A phase I trial in patients with adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef]
- Xing, P.; Michael, M.; Apostolopoulos, V.; Prenzoska, J.; Marshall, C.; Bishop, J.; McKenzie, I. Phase-I study of synthetic muc1 peptides in breast-cancer. Int. J. Oncol. 1995, 6, 1283–1289. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Tsibanis, A.; Tsikkinis, A.; Drakaki, H.; Loveland, B.E.; Piddlesden, S.J.; Plebanski, M.; Pouniotis, D.S.; Alexis, M.N.; et al. Pilot phase III immunotherapy study in early-stage breast cancer patients using oxidized mannan-MUC1 [ISRCTN71711835]. Breast Cancer Res. 2006, 8, R27. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Tsibanis, A.; Tsikkinis, A.; Stojanovska, L.; McKenzie, I.F.; Vassilaros, S. Dendritic cell immunotherapy: Clinical outcomes. Clin. Transl. Immunol. 2014, 3, e21. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Thalhammer, T.; Tzakos, A.G.; Stojanovska, L. Targeting antigens to dendritic cell receptors for vaccine development. J. Drug Deliv. 2013, 2013, 869718. [Google Scholar] [CrossRef]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef]
- Miller, S.; Senior, P.V.; Prakash, M.; Apostolopoulos, V.; Sakkal, S.; Nurgali, K. Leukocyte populations and IL-6 in the tumor microenvironment of an orthotopic colorectal cancer model. Acta Biochim. Biophys. Sin. 2016, 48, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Soliman, H.; Rawal, B.; Fulp, J.; Lee, J.H.; Lopez, A.; Bui, M.M.; Khalil, F.; Antonia, S.; Yfantis, H.G.; Lee, D.H.; et al. Analysis of indoleamine 2–3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol. Immunother. 2013, 62, 829–837. [Google Scholar] [CrossRef]
- Nagarajan, D.; McArdle, S.E.B. Immune Landscape of Breast Cancers. Biomedicines 2018, 6, 20. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef] [Green Version]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin. J. Cancer 2011, 30, 603–611. [Google Scholar] [CrossRef]
- Leo, C.; Cotic, C.; Pomp, V.; Fink, D.; Varga, Z. Overexpression of Lox in triple-negative breast cancer. Ann. Diagn. Pathol. 2018, 34, 98–102. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef]
- El-Haibi, C.P.; Bell, G.W.; Zhang, J.; Collmann, A.Y.; Wood, D.; Scherber, C.M.; Csizmadia, E.; Mariani, O.; Zhu, C.; Campagne, A.; et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc. Natl. Acad. Sci. USA 2012, 109, 17460–17465. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, A.; Nagel, A.; Szade, J.; Majewska, H.; Skokowski, J.; Seroczynska, B.; Stokowy, T.; Welnicka-Jaskiewicz, M.; Zaczek, A.J. Aggressive Phenotype of Cells Disseminated via Hematogenous and Lymphatic Route in Breast Cancer Patients. Transl. Oncol. 2018, 11, 722–731. [Google Scholar] [CrossRef]
- Qi, L.N.; Xiang, B.D.; Wu, F.X.; Ye, J.Z.; Zhong, J.H.; Wang, Y.Y.; Chen, Y.Y.; Chen, Z.S.; Ma, L.; Chen, J.; et al. Circulating Tumor Cells Undergoing EMT Provide a Metric for Diagnosis and Prognosis of Patients with Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4731–4744. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Whiteside, T. The Local Tumor Microenvironment, 1st ed.; Springer: Heidelberg, Germany, 2007; p. 503. [Google Scholar]
- Su, S.; Liao, J.; Liu, J.; Huang, D.; He, C.; Chen, F.; Yang, L.; Wu, W.; Chen, J.; Lin, L.; et al. Blocking the recruitment of naive CD4+ T cells reverses immunosuppression in breast cancer. Cell Res. 2017, 27, 461. [Google Scholar] [CrossRef]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.B.A.G.; van Vugt, M.A.T.M.; de Vries, E.G.E.; Schröder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Woo, E.Y.; Chu, C.S.; Goletz, T.J.; Schlienger, K.; Yeh, H.; Coukos, G.; Rubin, S.C.; Kaiser, L.R.; June, C.H. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001, 61, 4766–4772. [Google Scholar]
- Strauss, L.; Bergmann, C.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2007, 13, 6301–6311. [Google Scholar] [CrossRef]
- Dushyanthen, S.; Beavis, P.A.; Savas, P.; Teo, Z.L.; Zhou, C.; Mansour, M.; Darcy, P.K.; Loi, S. Relevance of tumor-infiltrating lymphocytes in breast cancer. BMC Med. 2015, 13, 202. [Google Scholar] [CrossRef]
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52, 178–188. [Google Scholar] [CrossRef]
- Devereaux, J.; Nurgali, K.; Kiatos, D.; Sakkal, S.; Apostolopoulos, V. Effects of platelet-rich plasma and platelet-poor plasma on human dermal fibroblasts. Maturitas 2018, 117, 34–44. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Buchsbaum, R.J.; Oh, S.Y. Breast Cancer-Associated Fibroblasts: Where we are and where we need to go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef]
- Martens, J.W.; Sieuwerts, A.M.; Bolt-deVries, J.; Bosma, P.T.; Swiggers, S.J.; Klijn, J.G.; Foekens, J.A. Aging of stromal-derived human breast fibroblasts might contribute to breast cancer progression. Thromb. Haemost. 2003, 89, 393–404. [Google Scholar]
- Palmieri, C.; Roberts-Clark, D.; Assadi-Sabet, A.; Coope, R.C.; O’Hare, M.; Sunters, A.; Hanby, A.; Slade, M.J.; Gomm, J.J.; Lam, E.W.; et al. Fibroblast growth factor 7, secreted by breast fibroblasts, is an interleukin-1beta-induced paracrine growth factor for human breast cells. J. Endocrinol. 2003, 177, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef]
- Tchou, J.; Kossenkov, A.V.; Chang, L.; Satija, C.; Herlyn, M.; Showe, L.C.; Puré, E. Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med. Genomics 2012, 5, 39. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Barnes, N.; Pietersz, G.A.; McKenzie, I.F. Ex vivo targeting of the macrophage mannose receptor generates anti-tumor CTL responses. Vaccine 2000, 18, 3174–3184. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Lofthouse, S.A.; Popovski, V.; Chelvanayagam, G.; Sandrin, M.S.; McKenzie, I.F. Peptide mimics of a tumor antigen induce functional cytotoxic T cells. Nat. Biotechnol. 1998, 16, 276–280. [Google Scholar] [CrossRef]
- Sheng, K.C.; Day, S.; Wright, M.D.; Stojanovska, L.; Apostolopoulos, V. Enhanced Dendritic Cell-Mediated Antigen-Specific CD4+ T Cell Responses: IFN-Gamma Aids TLR Stimulation. J. Drug Deliv. 2013, 2013, 516749. [Google Scholar] [CrossRef]
- Sheng, K.C.; Pouniotis, D.S.; Wright, M.D.; Tang, C.K.; Lazoura, E.; Pietersz, G.A.; Apostolopoulos, V. Mannan derivatives induce phenotypic and functional maturation of mouse dendritic cells. Immunology 2006, 118, 372–383. [Google Scholar] [CrossRef]
- Tang, C.K.; Lodding, J.; Minigo, G.; Pouniotis, D.S.; Plebanski, M.; Scholzen, A.; McKenzie, I.F.; Pietersz, G.A.; Apostolopoulos, V. Mannan-mediated gene delivery for cancer immunotherapy. Immunology 2007, 120, 325–335. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Alexandrow, M.G.; Moses, H.L. Transforming growth factor beta 1 inhibits mouse keratinocytes late in G1 independent of effects on gene transcription. Cancer Res. 1995, 55, 3928–3932. [Google Scholar]
- Obeid, E.; Nanda, R.; Fu, Y.X.; Olopade, O.I. The role of tumor-associated macrophages in breast cancer progression (review). Int. J. Oncol. 2013, 43, 5–12. [Google Scholar] [CrossRef]
- Klingen, T.A.; Chen, Y.; Aas, H.; Wik, E.; Akslen, L.A. Tumor-associated macrophages are strongly related to vascular invasion, non-luminal subtypes, and interval breast cancer. Hum. Pathol. 2017, 69, 72–80. [Google Scholar]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Pang, M.F.; Georgoudaki, A.M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef]
- Fuxe, J.; Karlsson, M.C. TGF-beta-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin. Cancer Biol. 2012, 22, 455–461. [Google Scholar] [CrossRef]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Gan, L.; Qiu, Z.; Huang, J.; Li, Y.; Huang, H.; Xiang, T.; Wan, J.; Hui, T.; Lin, Y.; Li, H.; et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway. Int. J. Biol. Sci. 2016, 12, 1533–1543. [Google Scholar] [Green Version]
- Ding, J.; Jin, W.; Chen, C.; Shao, Z.; Wu, J. Tumor Associated Macrophage × Cancer Cell Hybrids May Acquire Cancer Stem Cell Properties in Breast Cancer. PLoS ONE 2012, 7, e41942. [Google Scholar] [CrossRef]
- Brady, N.J.; Chuntova, P.; Schwertfeger, K.L. Macrophages: Regulators of the Inflammatory Microenvironment during Mammary Gland Development and Breast Cancer. Mediat. Inflamm. 2016, 2016, 13. [Google Scholar] [CrossRef]
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-associated macrophages in breast cancer: Distinct subsets, distinct functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef]
- Santarpia, M.; González-Cao, M.; Viteri, S.; Karachaliou, N.; Altavilla, G.; Rosell, R. Programmed cell death protein-1/programmed cell death ligand-1 pathway inhibition and predictive biomarkers: Understanding transforming growth factor-beta role. Transl. Lung Cancer Res. 2015, 4, 728–742. [Google Scholar]
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.-S.; Lee, E.S.; Jung, S.-Y.; Kwon, Y.; Kong, S.-Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730. [Google Scholar] [CrossRef] [Green Version]
- Treffers, L.W.; Hiemstra, I.H.; Kuijpers, T.W.; van den Berg, T.K.; Matlung, H.L. Neutrophils in cancer. Immunol. Rev. 2016, 273, 312–328. [Google Scholar] [CrossRef]
- Kerfoot, S.M.; Raharjo, E.; Ho, M.; Kaur, J.; Serirom, S.; McCafferty, D.M.; Burns, A.R.; Patel, K.D.; Kubes, P. Exclusive neutrophil recruitment with oncostatin M in a human system. Am. J. Pathol. 2001, 159, 1531–1539. [Google Scholar] [CrossRef]
- Modur, V.; Feldhaus, M.J.; Weyrich, A.S.; Jicha, D.L.; Prescott, S.M.; Zimmerman, G.A.; McIntyre, T.M. Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J. Clin. Investig. 1997, 100, 158–168. [Google Scholar] [CrossRef]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast Cancer Cells Stimulate Neutrophils to Produce Oncostatin M: Potential Implications for Tumor Progression. Cancer Res. 2005, 65, 8896. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Bankfalvi, A.; Gu, X.; Eberhardt, W.E.; Zeidler, R.; Lang, S.; Brandau, S. Neutrophils Activate Tumoral CORTACTIN to Enhance Progression of Orohypopharynx Carcinoma. Front. Immunol. 2013, 4, 33. [Google Scholar] [CrossRef] [Green Version]
- Iwase, T.; Sangai, T.; Sakakibara, M.; Sakakibara, J.; Ishigami, E.; Hayama, S.; Nakagawa, A.; Masuda, T.; Tabe, S.; Nagashima, T. An increased neutrophil-to-lymphocyte ratio predicts poorer survival following recurrence for patients with breast cancer. Mol. Clin. Oncol. 2017, 6, 266–270. [Google Scholar] [CrossRef]
- Soto-Perez-de-Celis, E.; Chavarri-Guerra, Y.; Leon-Rodriguez, E.; Gamboa-Dominguez, A. Tumor-Associated Neutrophils in Breast Cancer Subtypes. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 2689–2694. [Google Scholar]
- Jovanovic, B.; Beeler, J.S.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Ashby, W.J.; Lehmann, B.D.; Zijlstra, A.; Pietenpol, J.A.; Moses, H.L. Transforming growth factor beta receptor type III is a tumor promoter in mesenchymal-stem like triple negative breast cancer. Breast Cancer Res. 2014, 16, R69. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1“ versus “N2“ TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Lagraoui, M.; Gagnon, L. Enhancement of human neutrophil survival and activation by TGF-beta 1. Cell. Mol. Biol. (Noisy-le-grand) 1997, 43, 313–318. [Google Scholar]
- Perret, J.; McDonald, C.; Apostolopoulos, V. Elevated serum interleukin-5 levels in severe chronic obstructive pulmonary disease. Acta Biochim. Biophys. Sin. 2017, 49, 560–563. [Google Scholar] [CrossRef] [Green Version]
- Filippone, R.T.; Sahakian, L.; Apostolopoulos, V.; Nurgali, K. Eosinophils in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1140–1151. [Google Scholar] [CrossRef]
- Gouon-Evans, V.; Rothenberg, M.E.; Pollard, J.W. Postnatal mammary gland development requires macrophages and eosinophils. Development 2000, 127, 2269–2282. [Google Scholar]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Lucarini, V.; Marone, G.; Mattei, F.; Marone, G.; Schiavoni, G. Eosinophils: The unsung heroes in cancer? Oncoimmunology 2018, 7, e1393134. [Google Scholar] [CrossRef]
- Sakkal, S.; Miller, S.; Apostolopoulos, V.; Nurgali, K. Eosinophils in Cancer: Favourable or Unfavourable? Curr. Med. Chem. 2016, 23, 650–666. [Google Scholar] [CrossRef]
- Szalayova, G.; Ogrodnik, A.; Spencer, B.; Wade, J.; Bunn, J.; Ambaye, A.; James, T.; Rincon, M. Human breast cancer biopsies induce eosinophil recruitment and enhance adjacent cancer cell proliferation. Breast Cancer Res. Treat. 2016, 157, 461–474. [Google Scholar] [CrossRef] [Green Version]
- Amini, R.-M.; Aaltonen, K.; Nevanlinna, H.; Carvalho, R.; Salonen, L.; Heikkilä, P.; Blomqvist, C. Mast cells and eosinophils in invasive breast carcinoma. BMC Cancer 2007, 7, 165. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Fuchsberger, M.; Xiang, S.D.; Apostolopoulos, V.; Plebanski, M. Myeloid derived suppressor cells and their role in diseases. Curr. Med. Chem. 2013, 20, 1437–1444. [Google Scholar] [CrossRef]
- Tsai, C.S.; Chen, F.H.; Wang, C.C.; Huang, H.L.; Jung, S.M.; Wu, C.J.; Lee, C.C.; McBride, W.H.; Chiang, C.S.; Hong, J.H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- Ochoa, A.C.; Zea, A.H.; Hernandez, C.; Rodriguez, P.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721s–726s. [Google Scholar] [CrossRef]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Faour, I.; Kanbar, J.; Quadri, A.A.; Albashir, M.; Elkord, E. Myeloid cells in circulation and tumor microenvironment of breast cancer patients. Cancer Immunol. Immunother. 2017, 66, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Law, A.M.K.; Lim, E.; Ormandy, C.J.; Gallego-Ortega, D. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, R123–R144. [Google Scholar] [CrossRef] [Green Version]
- Bu, X.; Yao, Y.; Li, X. Immune Checkpoint Blockade in Breast Cancer Therapy. Adv. Exp. Med. Biol. 2017, 1026, 383–402. [Google Scholar]
- Walker, L.S.; Sansom, D.M. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015, 36, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Yu, H.; Yang, J.; Jiao, S.; Li, Y.; Zhang, W.; Wang, J. Cytotoxic T lymphocyte antigen 4 expression in human breast cancer: Implications for prognosis. Cancer Immunol. Immunother. 2015, 64, 853–860. [Google Scholar] [CrossRef]
- Hartkopf, A.D.; Taran, F.A.; Wallwiener, M.; Walter, C.B.; Krämer, B.; Grischke, E.M.; Brucker, S.Y. PD-1 and PD-L1 Immune Checkpoint Blockade to Treat Breast Cancer. Breast Care 2016, 11, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 2016, 9, 47. [Google Scholar] [CrossRef]
- Ren, X.; Wu, H.; Lu, J.; Zhang, Y.; Luo, Y.; Xu, Q.; Shen, S.; Liang, Z. PD1 protein expression in tumor infiltrated lymphocytes rather than PDL1 in tumor cells predicts survival in triple-negative breast cancer. Cancer Biol. Ther. 2018, 19, 373–380. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Birnbaum, D.; Mamessier, E. The PD1/PDL1 axis, a promising therapeutic target in aggressive breast cancers. Oncoimmunology 2016, 5, e1085148. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. Immunology at the maternal-fetal interface: Lessons for T cell tolerance and suppression. Annu. Rev. Immunol. 2000, 18, 367–391. [Google Scholar] [CrossRef]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. ImmunoTher. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.C.; Lan, J.L.; Chen, D.Y.; Yang, C.Y.; Chen, Y.M.; Li, J.P.; Huang, C.Y.; Liu, P.E.; Wang, X.; Tan, T.H. The kinase GLK controls autoimmunity and NF-kappaB signaling by activating the kinase PKC-theta in T cells. Nat. Immunol. 2011, 12, 1113–1118. [Google Scholar] [CrossRef]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Isla Larrain, M.T.; Rabassa, M.E.; Lacunza, E.; Barbera, A.; Cretón, A.; Segal-Eiras, A.; Croce, M.V. IDO is highly expressed in breast cancer and breast cancer-derived circulating microvesicles and associated to aggressive types of tumors by in silico analysis. Tumor Biol. 2014, 35, 6511–6519. [Google Scholar] [CrossRef]
- Asghar, K.; Loya, A.; Rana, I.A.; Tahseen, M.; Ishaq, M.; Farooq, A.; Bakar, M.A.; Masood, I. Indoleamine 2,3-dioxygenase expression and overall survival in patients diagnosed with breast cancer in Pakistan. Cancer Manag. Res. 2018, 11, 475–481. [Google Scholar] [CrossRef]
- Dill, E.A.; Dillon, P.M.; Bullock, T.N.; Mills, A.M. IDO expression in breast cancer: An assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod. Pathol. 2018, 31, 1513–1522. [Google Scholar] [CrossRef]
- Fraschilla, I.; Pillai, S. Viewing Siglecs through the lens of tumor immunology. Immunol. Rev. 2017, 276, 178–191. [Google Scholar] [CrossRef] [Green Version]
- Tanida, S.; Akita, K.; Ishida, A.; Mori, Y.; Toda, M.; Inoue, M.; Ohta, M.; Yashiro, M.; Sawada, T.; Hirakawa, K.; et al. Binding of the sialic acid-binding lectin, Siglec-9, to the membrane mucin, MUC1, induces recruitment of beta-catenin and subsequent cell growth. J. Biol. Chem. 2013, 288, 31842–31852. [Google Scholar] [CrossRef]
- Singh, P.K.; Hollingsworth, M.A. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 2006, 16, 467–476. [Google Scholar] [CrossRef]
- Workman, H.C.; Sweeney, C.; Carraway, K.L., 3rd. The membrane mucin Muc4 inhibits apoptosis induced by multiple insults via ErbB2-dependent and ErbB2-independent mechanisms. Cancer Res. 2009, 69, 2845–2852. [Google Scholar] [CrossRef]
- Gong, J.; Apostolopoulos, V.; Chen, D.; Chen, H.; Koido, S.; Gendler, S.J.; McKenzie, I.F.; Kufe, D. Selection and characterization of MUC1-specific CD8+ T cells from MUC1 transgenic mice immunized with dendritic-carcinoma fusion cells. Immunology 2000, 101, 316–324. [Google Scholar] [CrossRef]
- Koido, S.; Enomoto, Y.; Apostolopoulos, V.; Gong, J. Tumor regression by CD4 T-cells primed with dendritic/tumor fusion cell vaccines. Anticancer Res. 2014, 34, 3917–3924. [Google Scholar]
- Pietersz, G.A.; Li, W.; Popovski, V.; Caruana, J.A.; Apostolopoulos, V.; McKenzie, I.F. Parameters for using mannan-MUC1 fusion protein to induce cellular immunity. Cancer Immunol. Immunother. CII 1998, 45, 321–326. [Google Scholar] [CrossRef]
- Pietersz, G.A.; Pouniotis, D.S.; Apostolopoulos, V. Design of peptide-based vaccines for cancer. Curr. Med. Chem. 2006, 13, 1591–1607. [Google Scholar] [CrossRef]
- Son, H.Y.; Apostolopoulos, V.; Kim, C.W. T/Tn immunotherapy avoiding immune deviation. Int. J. Immunopathol. Pharmacol. 2016, 29, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Son, H.Y.; Apostolopoulos, V.; Kim, C.W. Mannosylated T/Tn with Freund’s adjuvant induces cellular immunity. Int. J. Immunopathol. Pharmacol. 2018, 31, 394632017742504. [Google Scholar] [CrossRef]
- Tang, C.K.; Katsara, M.; Apostolopoulos, V. Strategies used for MUC1 immunotherapy: Human clinical studies. Expert Rev. Vaccines 2008, 7, 963–975. [Google Scholar] [CrossRef]
- Macao, B.; Johansson, D.G.; Hansson, G.C.; Hard, T. Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin. Nat. Struct. Mol. Biol. 2006, 13, 71–76. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef]
- Huang, L.; Chen, D.; Liu, D.; Yin, L.; Kharbanda, S.; Kufe, D. MUC1 oncoprotein blocks glycogen synthase kinase 3beta-mediated phosphorylation and degradation of beta-catenin. Cancer Res. 2005, 65, 10413–10422. [Google Scholar] [CrossRef]
- Jing, X.; Liang, H.; Hao, C.; Yang, X.; Cui, X. Overexpression of MUC1 predicts poor prognosis in patients with breast cancer. Oncol. Rep. 2019, 41, 801–810. [Google Scholar] [CrossRef]
- Dong, R.; Wang, Q.; He, X.L.; Chu, Y.K.; Lu, J.G.; Ma, Q.J. Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-a-induced epithelial-mesenchymal transition of MCF-7 cells. Braz. J. Med. Biol. Res. 2007, 40, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.T.; Dwyer, R.M.; Kelly, J.; Khan, S.; Murphy, J.M.; Curran, C.; Miller, N.; Hennessy, E.; Dockery, P.; Barry, F.P.; et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 2010, 124, 317–326. [Google Scholar] [CrossRef]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; Kellokumpu-Lehtinen, P.-L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. BCR 2015, 17, 101. [Google Scholar] [CrossRef]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.-C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Ownby, H.E.; Roi, L.D.; Isenberg, R.R.; Brennan, M.J. Peripheral lymphocyte and eosinophil counts as indicators of prognosis in primary breast cancer. Cancer 1983, 52, 126–130. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. CII 2009, 58, 49–59. [Google Scholar] [CrossRef]
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389. [Google Scholar] [CrossRef]
- Chen, X.; Shao, Q.; Hao, S.; Zhao, Z.; Wang, Y.; Guo, X.; He, Y.; Gao, W.; Mao, H. CTLA-4 positive breast cancer cells suppress dendritic cells maturation and function. Oncotarget 2017, 8, 13703–13715. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, C.; Xian, J.; Zhang, M.; Cao, Y.; Cao, Y. Expression of programmed cell death protein 1 (PD-1) and indoleamine 2,3-dioxygenase (IDO) in the tumor microenvironment and in tumor-draining lymph nodes of breast cancer. Hum. Pathol. 2018, 75, 81–90. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.; Cho, M.S.; Lim, W.; Moon, B.-I.; Sung, S.H. Strong Correlation of Indoleamine 2,3-Dioxygenase 1 Expression with Basal-Like Phenotype and Increased Lymphocytic Infiltration in Triple-Negative Breast Cancer. J. Cancer 2017, 8, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, Y.; Morimatsu, M.; Nishijima, K.-I.; Usui, T.; Yamamoto, S.; Suyama, H.; Ozaki, K.; Ito, T.; Ono, E. A soluble form of Siglec-9 provides an antitumor benefit against mammary tumor cells expressing MUC1 in transgenic mice. Biochem. Biophys. Res. Commun. 2014, 450, 532–537. [Google Scholar] [CrossRef]
- Ohta, M.; Ishida, A.; Toda, M.; Akita, K.; Inoue, M.; Yamashita, K.; Watanabe, M.; Murata, T.; Usui, T.; Nakada, H. Immunomodulation of monocyte-derived dendritic cells through ligation of tumor-produced mucins to Siglec-9. Biochem. Biophys. Res. Commun. 2010, 402, 663–669. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogens and Its Receptors into Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Type | Mechanisms | Model | Detection | Ref |
|---|---|---|---|---|
| Epithelial-mesenchymal transition (EMT) (via transcription factors Snail, Twist-1 and Lox and markers vimentin and N-cadherin) | Involved in tumor progression and metastasis through signaling pathways such as TGFβ, NF-κB, Wnt, Notch | [60,61] | ||
| Lox important in extracellular matrix (ECM) → invasion and metastasis | Triple negative breast cancer (TNBC) and non-TNBC human samples | Immunohistochemistry (IHC) | [62] | |
| ↑Lox in TNBC | ||||
| NF-κB → ↑Snail → TNF-α induced EMT | MCF-7 Cell line | Cell migration (CM) | [166] | |
| Real time polymerase chain reaction (RTPCR) | ||||
| Western Blot (WB) | ||||
| MSC’s (Mesenchymal stem cells) ↑metastasis through facilitation of EMT | MDA-MB-231, T47D and SK-Br3 cell lines | Low-density array | [167] | |
| RT-PCR | ||||
| Gene expression and proliferation assays | ||||
| Immune cells Tumor-infiltrating Lymphocytes (TILs), T-cells (Tregs: CD4, CD25, FOXP3; CD8+; CD4+ Th cells) | Suppress T-cell proliferation | [74] | ||
| Induce tumor cell death via IFN-γ and granzyme-perforin molecules | ||||
| naïve CD4+ T cell recruitment → ↓Immunosuppression | MDA-MB-231 cell line | IHC and immunofluorescence staining, Flow cytometry (FC), Migration assay | [70] | |
| Primary breast carcinoma | ||||
| CD4+ naïve T cell | Female NOD/scid mice | qRT-PCR, Binding assays, WB | ||
| Humanized mice NOD/SCID/IL2rγnull (NSG) | ||||
| Tumor-associated | TAMs are re-programmed to inhibit lymphocyte functions through release of inhibitory cytokines such as IL-10, prostaglandins or reactive oxygen species (ROS) | [91,92] | ||
| macrophage (TAM) | ↑CD163+ in non-luminal and basal-like breast caner | Human tumor tissue | IHC with CD163 | [96] |
| breast cancer cell-secreted factors modulate macrophage differentiation to M2 status | Human tumor tissue | IHC | [168] | |
| FC | ||||
| Cell line MCF-7, MDA-MB231 and T47D | ELISA | |||
| Zymography | ||||
| ↑CD163+ in tumor stroma of TNBC | Human Luminal A and Triple Neg/basal-like tissue | IHC | [169] | |
| Gene Expression | ||||
| Cancer-associated Fibroblast (CAF) | Shown to secrete various growth factors and cytokines associated with promoting breast cancer proliferation | [79] | ||
| CAFs derived from Her2+ breast cancers → ↑actin cytoskeleton and integrin signaling | Breast tumors sub grouped according to receptor expression | IHC | [85] | |
| Gene Expression | ||||
| ER+ expressing CD146neg → ↑tumor resistance to tamoxifen | Human tissue (Stage II & III, ER+ and/or ER−) | Immunocytochemistry (ICC) | [170] | |
| ER+ expressing CD146pos → ↓tumor resistance to tamoxifen | MCF-7 cell line | Gene expression | ||
| TNBC exhibit CAF subsets, ↑CAF-S1 → ↑T Lymphocyte survival → ↑Treg → Ø T effector proliferation → Immunosuppression | Female BC patient cohort (Luminal, HER2 and TN subtype tissues) | FC | [171] | |
| IHC | ||||
| Tumor-associated Neutrophils (TANs) | N2 phenotype: pro-tumorigenic or pro-inflammatory | [172] | ||
| N1 phenotype: anti-tumorigenic | ||||
| Oncostastin M expressed by TANs → ↑angiogenesis and metastasis | MDA-MB-231 & T47D cell lines | ICC | [112] | |
| ELISA | ||||
| ↑TAN in TNBC | Stage I-III breast cancer patient tumors divided into three subtypes: hormone-receptor [HR]-positive, HER2-negative (HR+, HER2-ve); HER2-positive and triple negative (TN) | Hematoxylin & eosin | [115] | |
| IHC | ||||
| ↑TβRIII (TGF-β receptor) in TNBC → ↑mesenchymal-stem like (MSL) TNBC cells → cell migration, invasion, and tumorigenicity | MSL cell lines SUM159, MDA-MB-231 and MDA-MB-157 | Cell proliferation assay | [116] | |
| CM and invasion assay | ||||
| Immunoblotting | ||||
| FC | ||||
| Gene Expression | ||||
| Tumor-associated Eosinophils | High presence of eosinophils at biopsy site may be linked to proliferation rate of tumor cells adjacent to wound | Female patients with primary breast cancer | Peripheral eosinophil counts | [173] |
| Myeloid-derived suppressor cells (MDSCs) | ↑Arginase 1 (ARG1) + nitric oxide synthase (iNOS) → ↑superoxide and nitric oxide (NO) → Ø lymphocyte responses → ↑iNOS in surrounding cells → ↑tumor growth and ↓ immune cell functions | [127,128] | ||
| stage IV patients with extensive metastasis → ↑MDSC | Blood from patients with stages I–IV solid malignancies obtained prior to surgery | FC | [174] | |
| ↑MDSC correlates with worse prognosis | Peripheral blood specimens stage IV breast cancer patients | FC | [175] | |
| Proliferation assay | ||||
| MDSC ↑IDO → ↓tryptophan → Ø T-cell proliferation and induced T-cell apoptosis | Female breast cancer patients (Stages I–III) | IHC | [176] | |
| RT-PCR | ||||
| WB | ||||
| ELISA |
| Immune Checkpoint Factor | Mechanisms | Model | Detection | Ref |
|---|---|---|---|---|
| CTLA4 | Expressed on the surface of activated T-cells and a subset of Tregs ↓T-cell activation → Anti-T cell response | [177] | ||
| CTLA4 blockade → Ø proliferation and induced apoptosis of CTLA-4+ breast cancer cells | MDA-MB-231, SKBR3, MCF-7 and T47D cell lines | FC | [178] | |
| WB | ||||
| ↑CTLA-4 in lymphocytes → better prognosis | 130 BC patients | IHC | [134] | |
| ↑CTLA-4 in T cells → worse prognosis | ||||
| PD-1 (PD-L1/PD-L2) | PD-1 is expressed by activated lymphocytes → ligation PD-L1/PD-L2 → ↓T-cell activity → poor prognosis | [135] | ||
| TNBC subtype; ↑PD-L1 → suppresses auto-immunity—T cell proliferation—Cytokine production—Cytotoxic activity | [136] | |||
| PD1 ↑TILs, but ↓PDL1 in T cells → positive TNBC prognostic factor | negative ER, PR, and HER-2 BC patients | IHC | [137] | |
| RNAscope | ||||
| ↑PD-L1 in Basal & TNBC subtypes → ↑cytotoxic local immune response → ↑better survival | BC patient is Cell lines | [138] | ||
| Indoleamine 2,3-dioxygenase (IDO) | Catalyzes the oxidative break-down of tryptophan via kynurenine pathway in the presence of IFN-γ → enabling immune escape | [130] | ||
| Correlation between expression of IDO and PD-1 in myoepithelial, stromal, and T cells | Human BC patient tissues and healthy tissues | IHC | [179] | |
| WB | ||||
| RT-PCR | ||||
| ↑IDO expression in TNBC and basal-like BC | 200 TNBC patients | IHC | [180] | |
| RT-PCR | ||||
| IDO expression ↑ER+ as compared to ER− is | breast cancer tissue sections | IHC | [52] | |
| Siglec-9 | Siglec-9 found on neutrophils and Siglec-7 found on NK cells have been associated with anti-T immunity | [151] | ||
| Siglec-9 → T-associated MUC1 downstream signal transduction, following T cell proliferation | Transgenic mice and murine mammary T cell | WB, Immunoperoxidase staining, IHC | [181] | |
| Gene expression | ||||
| Proliferation (MTT) assay | ||||
| ↑Siglec-9 on DCs involved in immunoregulation through ligation with mucins in epithelial cancer | Human colon cancer cell line (LS 180 cells) | FC | [182] | |
| RT-PCR | ||||
| ELISA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barriga, V.; Kuol, N.; Nurgali, K.; Apostolopoulos, V. The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer. Cancers 2019, 11, 1205. https://doi.org/10.3390/cancers11081205
Barriga V, Kuol N, Nurgali K, Apostolopoulos V. The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer. Cancers. 2019; 11(8):1205. https://doi.org/10.3390/cancers11081205
Chicago/Turabian StyleBarriga, Vanessa, Nyanbol Kuol, Kulmira Nurgali, and Vasso Apostolopoulos. 2019. "The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer" Cancers 11, no. 8: 1205. https://doi.org/10.3390/cancers11081205
APA StyleBarriga, V., Kuol, N., Nurgali, K., & Apostolopoulos, V. (2019). The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer. Cancers, 11(8), 1205. https://doi.org/10.3390/cancers11081205