Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Angiogenic Switch
3. Cancer Immune-Mediated Dormancy
4. Metabolic Reprogramming
5. ECM Remodeling
6. Cancer Stem Cells (CSCs)
7. Epigenetic Modification
8. Noncoding RNAs (miRNAs)
9. Stress-Induced p38 Signaling
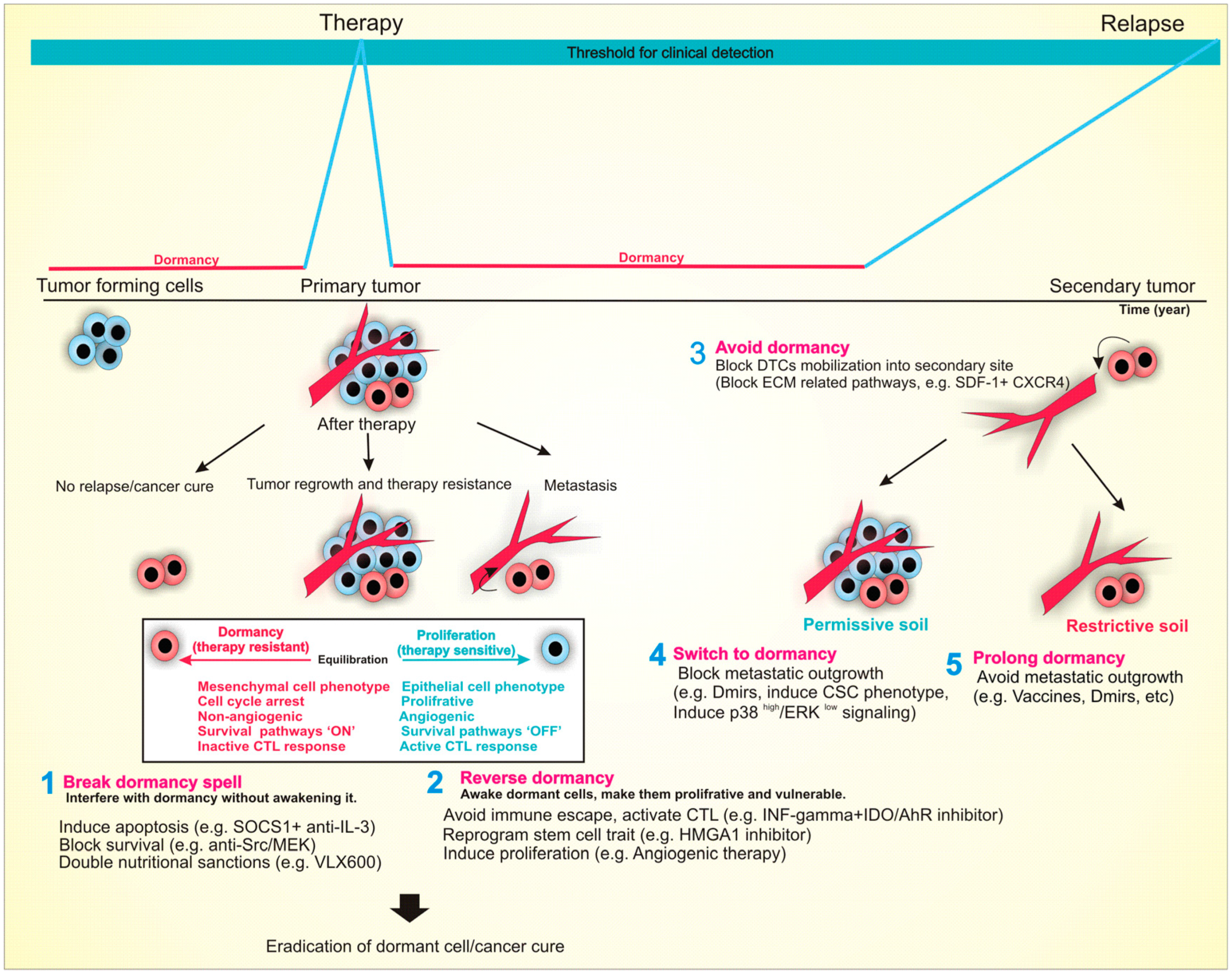
10. Conclusions and Clinical Implications
Funding
Acknowledgments
Conflicts of Interest
References
- Jahanban-Esfahlan, R.; de la Guardia, M.; Ahmadi, D.; Yousefi, B. Modulating tumor hypoxia by nanomedicine for effective cancer therapy. J. Cell. Physiol. 2017, 233, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, T. Tumor dormancy of primary and secondary cancers. Apmis: Acta Pathol. Microbiol. Immunol. Scand. 2008, 116, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Ya-Fei, D.; Juan-Juan, X. Tumor Cell Dormancy: How It Performs in Drug Resistance and Relapse. Prog. Biochem. Biophys. 2018, 45, 460–470. [Google Scholar]
- Hedley, B.D.; Chambers, A.F. Tumor dormancy and metastasis. Adv. Cancer Res. 2009, 102, 67–101. [Google Scholar] [PubMed]
- Manjili, M.H. Tumor dormancy and relapse: From a natural byproduct of evolution to a disease state. Cancer Res. 2017, 77, 2564–2569. [Google Scholar] [CrossRef] [PubMed]
- Paez, D.; Labonte, M.J.; Bohanes, P.; Zhang, W.; Benhanim, L.; Ning, Y.; Wakatsuki, T.; Loupakis, F.; Lenz, H.J. Cancer dormancy: A model of early dissemination and late cancer recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Khoon, M.C.S. Experimental models of bone metastasis: Opportunities for the study of cancer dormancy. Adv. Drug Deliv. Rev. 2015, 94, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Uhr, J.W.; Scheuermann, R.H.; Street, N.E.; Vitetta, E.S. Cancer dormancy: Opportunities for new therapeutic approaches. Nat. Med. 1997, 3, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Kumar, M.P.; Wheeler, S.E.; Young, C.L.; Venkataramanan, R.; Stolz, D.B.; Griffith, L.G.; Lauffenburger, D.A.; Wells, A. A Model of Dormant-Emergent Metastatic Breast Cancer Progression Enabling Exploration of Biomarker Signatures. Mol. Cell. Proteom. 2018, 17, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrios, J.; Wieder, R. Dual FGF-2, and integrin alpha5beta1 signaling mediate GRAF-induced RhoA inactivation in a model of breast cancer dormancy. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2009, 2, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fryknas, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Pahlman, S.; Schughart, L.A.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumor cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [PubMed]
- Torrano, V.; Carracedo, A. Quiescence-like Metabolism to Push Cancer Out of the Race. Cell Metab. 2017, 25, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Nur Saidy, N.R.; Collins, C.C.; Wang, Y. The epigenetic/noncoding origin of tumor dormancy. Trends Mol. Med. 2015, 21, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bloj, B.; Moses, C.; Sgro, A.; Plani-Lam, J.; Arooj, M.; Duffy, C.; Thiruvengadam, S.; Sorolla, A.; Rashwan, R.; Mancera, R.L.; et al. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget 2016, 7, 60535–60554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartosh, T.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [PubMed]
- Almog, N.; Briggs, C.; Beheshti, A.; Ma, L.; Wilkie, K.P.; Rietman, E.; Hlatky, L. Transcriptional changes induced by the tumor dormancy-associated microRNA-190. Transcription 2013, 4, 177–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control the dormant residual disease. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Liu, Y.; Di Giandomenico, S.; Bae, N.; Ndiaye-Lobry, D.; Deblasio, A.; Menendez, S.; Antipin, Y.; Reva, B.; Wevrick, R.; et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood 2012, 120, 1601–1612. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W. Chronic inflammation and breast cancer recurrence. J. Clin. Oncol. 2009, 27, 3418–3419. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-L.; Zhang, M.; Tang, Y.-L.; Liang, X.-H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. OncoTargets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-H.V.; Middleton, K.; You, L.; Sun, Y. A review of microfluidic approaches for investigating cancer extravasation during metastasis. Microsyst. Nanoeng. 2018, 4, 17104. [Google Scholar] [CrossRef] [Green Version]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Skelley, A.M.; Merdek, K.D.; Sprott, K.M.; Jiang, C.; Pierceall, W.E.; Lin, J.; Stocum, M.; Carney, W.P.; Smirnov, D.A. Microfluidics and Circulating Tumor Cells. J. Mol. Diagn. 2013, 15, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kim, J.; Song, H.; Sohn, K.Y.; Jeon, M.; Han, K.H. Microfluidic technologies for circulating tumor cell isolation. Analyst 2018, 143, 2936–2970. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Kondapaneni, R.V.; Narkhede, A.A. Bioengineered models to study tumor dormancy. J. Biol. Eng. 2019, 13, 3. [Google Scholar] [CrossRef]
- Pradhan, S.; Slater, J.H. Datasets describing hydrogel properties and cellular metrics for modeling of tumor dormancy. Data Brief 2019, 25, 104128. [Google Scholar] [CrossRef]
- Pradhan, S.; Sperduto, J.L.; Farino, C.J.; Slater, J.H. Engineered In Vitro Models of Tumor Dormancy and Reactivation. J. Biol. Eng. 2018, 12, 37. [Google Scholar] [CrossRef]
- Ambs, S.; Dennis, S.; Fairman, J.; Wright, M.; Papkoff, J. Inhibition of tumor growth correlates with the expression level of a human angiostatin transgene in transfected B16F10 melanoma cells. Cancer Res. 1999, 59, 5773–5777. [Google Scholar] [PubMed]
- Rofstad, E.K.; Graff, B.A. Thrombospondin-1-mediated metastasis suppression by the primary tumor in human melanoma xenografts. J. Investig. Dermatol. 2001, 117, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Bayko, L.; Rak, J.; Man, S.; Bicknell, R.; Ferrara, N.; Kerbel, R.S. The dormant in vivo phenotype of early-stage primary human melanoma: Termination by overexpression of vascular endothelial growth factor. Angiogenesis 1998, 2, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; Rak, J.W.; Klement, G.; Kerbel, R.S. Vascular endothelial growth factor isoform expression as a determinant of blood vessel patterning in human melanoma xenografts. Cancer Res. 2002, 62, 1838–1846. [Google Scholar] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumor dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; et al. Transcriptional switch of dormant tumors to the fast-growing angiogenic phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.L.; Pedersen, A.N.; Kantor, J.; Geisinger, K.; Long, R.; Zbieranski, N.; Townsend, A.; Shelton, B.; Brunner, N.; Kute, T.E. Relationship of nm23 to proteolytic factors, proliferation and motility in breast cancer tissues and cell lines. Br. J. Cancer 1998, 78, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, S.; Zhan, R.; Chaudhuri, A.; Watabe, M.; Pai, S.K.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Takano, Y.; et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 2006, 12, 933–938. [Google Scholar] [CrossRef]
- Seraj, M.J.; Samant, R.S.; Verderame, M.F.; Welch, D.R. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000, 60, 2764–2769. [Google Scholar]
- Straume, O.; Shimamura, T.; Lampa, M.J.; Carretero, J.; Oyan, A.M.; Jia, D.; Borgman, C.L.; Soucheray, M.; Downing, S.R.; Short, S.M.; et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 8699–8704. [Google Scholar] [CrossRef] [Green Version]
- Romero, I.; Garrido, F.; Garcia-Lora, A.M. Metastases in immune-mediated dormancy: A new opportunity for targeting cancer. Cancer Res. 2014, 74, 6750–6757. [Google Scholar] [CrossRef]
- Quesnel, B. Tumor dormancy and immunoescape. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2008, 116, 685–694. [Google Scholar] [CrossRef]
- Walker, N.D.; Elias, M.; Guiro, K.; Bhatia, R.; Greco, S.J.; Bryan, M.; Gergues, M.; Sandiford, O.A.; Ponzio, N.M.; Leibovich, S.J.; et al. Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis. 2019, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Feng, X.X.; Luo, C.; Wang, Y.; Li, D.; Shu, Y.; Wang, S.S.; Qin, J.; Li, Y.C.; Zou, J.M.; et al. 14,15-EET induces the infiltration and tumor-promoting function of neutrophils to trigger the growth of minimal dormant metastases. Oncotarget 2016, 7, 43324–43336. [Google Scholar] [CrossRef]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation triggers Zeb1-dependent to escape from tumor latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef]
- Saudemont, A.; Hamrouni, A.; Marchetti, P.; Liu, J.; Jouy, N.; Hetuin, D.; Colucci, F.; Quesnel, B. Dormant tumor cells develop cross-resistance to apoptosis induced by CTLs or imatinib mesylate via methylation of suppressor of cytokine signaling 1. Cancer Res. 2007, 67, 4491–4498. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X.; et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 2017, 8, 15207. [Google Scholar] [CrossRef]
- Keckesova, Z.; Donaher, J.L.; De Cock, J.; Freinkman, E.; Lingrell, S.; Bachovchin, D.A.; Bierie, B.; Tischler, V.; Noske, A.; Okondo, M.C.; et al. LACTB is a tumor suppressor that modulates lipid metabolism and cell state. Nature 2017, 543, 681–686. [Google Scholar] [CrossRef]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef] [Green Version]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K.; Bossler, F.; Lohrey, C.; Bulkescher, J.; Rosl, F.; Jansen, L.; Mayer, A.; Vaupel, P.; Durst, M.; Hoppe-Seyler, F. Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E990–E998. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Miele, M.E.; Hicks, D.J.; Phillips, K.K.; Trent, J.M.; Weissman, B.E.; Welch, D.R. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl. Cancer Inst. 1996, 88, 1731–1737. [Google Scholar] [CrossRef]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef]
- Allgayer, H.; Aguirre-Ghiso, J.A. The urokinase receptor (u-PAR)—A link between tumor cell dormancy and minimal residual disease in bone marrow? APMIS Acta Pathol. Microbiol. Immunol. Scand. 2008, 116, 602–614. [Google Scholar] [CrossRef]
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2014, 124, 156–168. [Google Scholar] [CrossRef]
- Rajbhandari, N.; Lin, W.C.; Wehde, B.L.; Triplett, A.A.; Wagner, K.U. Autocrine IGF1 signaling mediates pancreatic tumor cell dormancy in the absence of oncogenic drivers. Cell Rep. 2017, 18, 2243–2255. [Google Scholar] [CrossRef]
- Ruppender, N.; Larson, S.; Lakely, B.; Kollath, L.; Brown, L.; Coleman, I.; Coleman, R.; Nguyen, H.; Nelson, P.S.; Corey, E.; et al. Cellular adhesion promotes prostate cancer cells escape from dormancy. PLoS ONE 2015, 10, e0130565. [Google Scholar] [CrossRef]
- Mao, W.; Peters, H.L.; Sutton, M.N.; Orozco, A.F.; Pang, L.; Yang, H.; Lu, Z.; Bast, R.C., Jr. The role of vascular endothelial growth factor, interleukin 8, and insulinlike growth factor in sustaining autophagic DIRAS3-induced dormant ovarian cancer xenografts. Cancer 2019, 125, 1267–1280. [Google Scholar] [CrossRef]
- Yang, L.Y.; Shan, Y.M.; Zhang, Y.; Zhou, E.H.; Chen, X.P.; Zhang, H. Aurora kinase A induces chemotherapy resistance through revival of dormant cells in laryngeal squamous cell carcinoma. Head Neck 2019, 41, 2239–2248. [Google Scholar] [CrossRef]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2019, 216, 428–449. [Google Scholar]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbetaRIII-p38MAPK-pS249/T252RB pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra373. [Google Scholar] [CrossRef]
- Luo, X.L.; Deng, C.; Su, X.D.; Wang, F.; Chen, Z.; Wu, X.P.; Liang, S.B.; Liu, J.; Fu, L. Loss of MED12 induces tumor dormancy in human epithelial ovarian cancer via downregulation of EGFR. Cancer Res. 2018, 78, 3532–3543. [Google Scholar] [CrossRef] [Green Version]
- Sinha, G.; Rameshwar, P. N-cadherin in cancer dormancy. Cell Death 2015, 1, 23–27. [Google Scholar] [CrossRef]
- Abravanel, D.L.; Belka, G.K.; Pan, T.C.; Pant, D.K.; Collins, M.A.; Sterner, C.J.; Chodosh, L.A. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J. Clin. Investig. 2015, 125, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Marusawa, H.; Chiba, T. A marker for dormant cancer stem cells in human hepatocellular carcinoma. Gastroenterology 2011, 140, 1353–1355, discussion 1355. [Google Scholar] [CrossRef]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP Inhibitor Coco Reactivates Breast Cancer Cells at Lung Metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.K.; Balaji, K.C.; Watabe, K. Secreted Protein Acidic and Rich in Cysteine (SPARC) Mediates Metastatic Dormancy of Prostate Cancer in Bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Yang, F.; Huang, W.; Zhang, Y.; Bao, B.; Li, K.; Wei, F.; Zhang, C.; Jia, H. Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics 2019, 9, 424–435. [Google Scholar] [CrossRef]
- Shah, S.N.; Cope, L.; Poh, W.; Belton, A.; Roy, S.; Talbot, C.C., Jr.; Sukumar, S.; Huso, D.L.; Resar, L.M. HMGA1, a master regulator of tumor progression in triple-negative breast cancer cells. PLoS ONE 2013, 8, e63419. [Google Scholar] [CrossRef]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef]
- Jiang, J.; Zheng, M.; Zhang, M.; Yang, X.; Li, L.; Wang, S.-S.; Wu, J.-S.; Yu, X.-H.; Wu, J.-B.; Pang, X.; et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma Through miR-642b-3p. Neoplasia 2019, 21, 216–229. [Google Scholar] [CrossRef]
- Gregoire, J.M.; Fleury, L.; Salazar-Cardozo, C.; Alby, F.; Masson, V.; Arimondo, P.B.; Ausseil, F. Identification of epigenetic factors regulating the mesenchyme to epithelium transition by RNA interference screening in breast cancer cells. BMC Cancer 2016, 16, 700. [Google Scholar] [CrossRef]
- Brien, G.L.; Healy, E.; Jerman, E.; Conway, E.; Fadda, E.; O’Donovan, D.; Krivtsov, A.V.; Rice, A.M.; Kearney, C.J.; Flaus, A.; et al. A chromatin-independent role of Polycomb-like 1 to stabilize p53 and promote cellular quiescence. Genes Dev. 2015, 29, 2231–2243. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef]
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; Del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER(+) breast cancer. Nat. Cell Biol. 2018, 20, 211–221. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef]
- Tiram, G.; Segal, E.; Krivitsky, A.; Shreberk-Hassidim, R.; Ferber, S.; Ofek, P.; Udagawa, T.; Edry, L.; Shomron, N.; Roniger, M.; et al. Identification of dormancy-associated MicroRNAs for the design of osteosarcoma-targeted dendritic polyglycerol nanopolyplexes. ACS Nano 2016, 10, 2028–2045. [Google Scholar] [CrossRef]
- Nabavi, N.; Saidy, N.R.N.; Venalainen, E.; Haegert, A.; Parolia, A.; Xue, H.; Wang, Y.; Wu, R.; Dong, X.; Collins, C.; et al. miR-100-5p inhibition induces apoptosis in dormant prostate cancer cells and prevents the emergence of castration-resistant prostate cancer. Sci. Rep. 2017, 7, 4079. [Google Scholar] [CrossRef] [Green Version]
- Watson, K.L.; Jones, R.A.; Anthony, B.; Moorehead, R.A. The miR-200b/200a/429 cluster prevents metastasis and induces dormancy in a murine claudin-low mammary tumor cell line. Exp. Cell Res. 2018, 369, 17–36. [Google Scholar] [CrossRef]
- Maroni, P.; Bendinelli, P.; Matteucci, E.; Desiderio, M.A. The therapeutic effect of miR-125b is enhanced by the prostaglandin endoperoxide synthase 2/cyclooxygenase 2 blockade and hampers ETS1 in the context of the microenvironment of bone metastasis. Cell Death Dis. 2018, 9, 472. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Adam, A.P.; Aguirre-Ghiso, J.A. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle 2006, 5, 1799–1807. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell. Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef]
- Lotan, T.; Hickson, J.; Souris, J.; Huo, D.; Taylor, J.; Li, T.; Otto, K.; Yamada, S.D.; Macleod, K.; Rinker-Schaeffer, C.W. c-Jun NH2-terminal kinase activating kinase 1/mitogen-activated protein kinase kinase 4-mediated inhibition of SKOV3ip.1 ovarian cancer metastasis involves growth arrest and p21 up-regulation. Cancer Res. 2008, 68, 2166–2175. [Google Scholar] [CrossRef]
- Vander Griend, D.J.; Kocherginsky, M.; Hickson, J.A.; Stadler, W.M.; Lin, A.; Rinker-Schaeffer, C.W. Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005, 65, 10984–10991. [Google Scholar] [CrossRef]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.Y.; Sampson, D.; Flynn, E.; Watnick, R.S.; Straume, O.; Akslen, L.A.; Folkman, J.; et al. A model of human tumor dormancy: An angiogenic switch from the nonangiogenic phenotype. J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar] [CrossRef]
- Naumov, G.N.; Folkman, J.; Straume, O. Tumor dormancy due to failure of angiogenesis: Role of the microenvironment. Clin. Exp. Metastasis 2009, 26, 51–60. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, R.; Seidi, K.; Banimohamad-Shotorbani, B.; Jahanban-Esfahlan, A.; Yousefi, B. Combination of nanotechnology with vascular targeting agents for effective cancer therapy. J. Cell. Physiol. 2017, 233, 2982–2992. [Google Scholar] [CrossRef]
- Benzekry, S.; Gandolfi, A.; Hahnfeldt, P. Global dormancy of metastases due to systemic inhibition of angiogenesis. PLoS ONE 2014, 9, e84249. [Google Scholar] [CrossRef]
- Almog, N.; Henke, V.; Flores, L.; Hlatky, L.; Kung, A.L.; Wright, R.D.; Berger, R.; Hutchinson, L.; Naumov, G.N.; Bender, E.; et al. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 947–949. [Google Scholar] [CrossRef]
- Prunier, C.; Baker, D.; ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef]
- Shah, S.A.; Zarei, M.; Manjili, S.H.; Guruli, G.; Wang, X.Y.; Manjili, M.H. Immunotherapy of cancer: Targeting cancer during active disease or during dormancy? Immunotherapy 2017, 9, 943–949. [Google Scholar] [CrossRef]
- Baxevanis, C.N. T-cell recognition of non-mutated tumor antigens in healthy individuals: Connecting endogenous immunity and tumor dormancy. Cancer Immunol. Immunother. 2019, 68, 705–707. [Google Scholar] [CrossRef]
- Teng, M.W.; Swann, J.B.; Koebel, C.M.; Schreiber, R.D.; Smyth, M.J. Immune-mediated dormancy: An equilibrium with cancer. J. Leukoc. Biol. 2008, 84, 988–993. [Google Scholar] [CrossRef]
- Romero, I.; Garrido, C.; Algarra, I.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. T lymphocytes restrain spontaneous metastases in permanent dormancy. Cancer Res. 2014, 74, 1958–1968. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Saudemont, A.; Jouy, N.; Hetuin, D.; Quesnel, B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood 2005, 105, 2428–2435. [Google Scholar] [CrossRef]
- Manjili, M.H.; Butler, S.E. Role of Tregs in cancer dormancy or recurrence. Immunol. Investig. 2016, 45, 759–766. [Google Scholar] [CrossRef]
- Lan, Q.; Peyvandi, S.; Duffey, N.; Huang, Y.-T.; Barras, D.; Held, W.; Richard, F.; Delorenzi, M.; Sotiriou, C.; Desmedt, C.; et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2019, 38, 2814–2829. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- Aqbi, H.F.; Wallace, M.; Sappal, S.; Payne, K.K.; Manjili, M.H. IFN-gamma orchestrates tumor elimination, tumor dormancy, tumor escape, and progression. J. Leukoc. Biol. 2018, 103, 1219–1223. [Google Scholar] [CrossRef]
- Bakacs, T.; Mehrishi, J.N. Breast and other cancer dormancy as a therapeutic endpoint: Speculative recombinant T cell receptor ligand (RTL) adjuvant therapy worth considering? BMC Cancer 2010, 10, 251. [Google Scholar] [CrossRef]
- Minas, T.Z.; Han, J.; Javaheri, T.; Hong, S.H.; Schlederer, M.; Saygideger-Kont, Y.; Celik, H.; Mueller, K.M.; Temel, I.; Ozdemirli, M.; et al. YK-4-279 effectively antagonizes EWS-FLI1 induced leukemia in a transgenic mouse model. Oncotarget 2015, 6, 37678–37694. [Google Scholar] [CrossRef]
- Javaheri, T.; Kazemi, Z.; Pencik, J.; Pham, H.T.T.; Kauer, M.; Noorizadeh, R.; Sax, B.; Nivarthi, H.; Schlederer, M.; Maurer, B.; et al. Increased survival and cell cycle progression pathways are required for EWS/FLI1-induced malignant transformation. Cell Death Disease 2016, 7, e2419. [Google Scholar] [CrossRef]
- Morecki, S.; Pugatsch, T.; Levi, S.; Moshel, Y.; Slavin, S. Tumor-cell vaccination induces tumor dormancy in a murine model of B-cell leukemia/lymphoma (BCL1). Int. J. Cancer 1996, 65, 204–208. [Google Scholar] [CrossRef]
- Retsky, M.; Demicheli, R.; Hrushesky, W.; Baum, M.; Gukas, I. Surgery triggers outgrowth of latent distant disease in breast cancer: An inconvenient truth? Cancers 2010, 2, 305–337. [Google Scholar] [CrossRef]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.R.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10, eaan3464. [Google Scholar] [CrossRef]
- Mima, S.; Kakinuma, C.; Higuchi, T.; Saeki, K.; Yamada, T.; Uematsu, R.; Ishino, M.; Kito, N.; Nishikawa, H.; Kuniyoshi, H.; et al. FF-10502, an antimetabolite with novel activity on dormant cells, is superior to gemcitabine for targeting pancreatic cancer cells. J. Pharmacol. Exp. Ther. 2018, 366, 125–135. [Google Scholar] [CrossRef]
- Aqbi, H.F.; Tyutyunyk-Massey, L.; Keim, R.C.; Butler, S.E.; Thekkudan, T.; Joshi, S.; Smith, T.M.; Bandyopadhyay, D.; Idowu, M.O.; Bear, H.D.; et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018, 9, 22113–22122. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Green, J.E.; Chambers, A.F. Extracellular matrix: A gatekeeper in the transition from dormancy to metastatic growth. Eur. J. Cancer 2010, 46, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Keeratichamroen, S.; Lirdprapamongkol, K.; Svasti, J. Mechanism of ECM-induced dormancy and chemoresistance in A549 human lung carcinoma cells. Oncol. Rep. 2018, 39, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389. [Google Scholar] [CrossRef]
- Takeishi, S.; Nakayama, K.I. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 2016, 107, 875–881. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Heydarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Release 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Majidinia, M.; Aghazadeh, J.; Jahanban-Esfahlani, R.; Yousefi, B. The roles of Wnt/beta-catenin pathway in tissue development and regenerative medicine. J. Cell. Physiol. 2018, 233, 5598–5612. [Google Scholar] [CrossRef]
- Ford, A.M.; Mansur, M.B.; Furness, C.L.; van Delft, F.W.; Okamura, J.; Suzuki, T.; Kobayashi, H.; Kaneko, Y.; Greaves, M. Protracted dormancy of pre-leukemic stem cells. Leukemia 2015, 29, 2202–2207. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Roato, I.; Ferracini, R. Cancer Stem Cells, Bone and Tumor Microenvironment: Key Players in Bone Metastases. Cancers 2018, 10, 56. [Google Scholar] [CrossRef]
- Borgen, E.; Rypdal, M.C.; Sosa, M.S.; Renolen, A.; Schlichting, E.; Lønning, P.E.; Synnestvedt, M.; Aguirre-Ghiso, J.A.; Naume, B. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 2018, 20, 120. [Google Scholar] [CrossRef]
- Selli, C.; Turnbull, A.K.; Pearce, D.A.; Li, A.; Fernando, A.; Wills, J.; Renshaw, L.; Thomas, J.S.; Dixon, J.M.; Sims, A.H. Molecular changes during extended neoadjuvant letrozole treatment of breast cancer: Distinguishing acquired resistance from dormant tumours. Breast Cancer Res. 2019, 21, 2. [Google Scholar] [CrossRef]
- Majidinia, M.; Darband, S.G.; Kaviani, M.; Nabavi, S.M.; Jahanban-Esfahlan, R.; Yousefi, B. Cross-regulation between Notch signaling pathway and miRNA machinery in cancer. DNA Repair 2018, 66, 30–41. [Google Scholar] [CrossRef]
- Marlow, R.; Honeth, G.; Lombardi, S.; Cariati, M.; Hessey, S.; Pipili, A.; Mariotti, V.; Buchupalli, B.; Foster, K.; Bonnet, D.; et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 2013, 73, 6886–6899. [Google Scholar] [CrossRef]
- Almog, N.; Ma, L.; Schwager, C.; Brinkmann, B.G.; Beheshti, A.; Vajkoczy, P.; Folkman, J.; Hlatky, L.; Abdollahi, A. Consensus micro RNAs governing the switch of dormant tumors to the fast-growing angiogenic phenotype. PLoS ONE 2012, 7, e44001. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Adam, A.P.; Zhang, L.; Aguirre-Ghiso, J.A. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: An adaptive advantage for metastatic cells? Cancer Biol. Ther. 2006, 5, 729–735. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Dormancy Factor | Mode | Major Findings | Ref |
|---|---|---|---|
| Angiostatin | Inducer of angiogenic dormancy | Upregulation of Angiostatin drive long-term dormancy of primary tumors, inhibit tumor growth, and reduce cancer metastases. | [31] |
| Thrombospondin-1 | Inducer of angiogenic dormancy | Overexpression of Thrombospondin-1 inhibits melanoma angiogenesis, lung colonization, and spontaneous pulmonary metastasis. | [32] |
| VEGF/VPF121 | Inhibitor of angiogenic dormancy | Overexpression of VEGF/VPF121 result in tumor growth and escape from dormancy. | [33] |
| (VEGF(121) VEGF(165) overexpression | Inhibitor of angiogenic dormancy | The level and VEGF isoforms determine the fate of aggressive tumor growth vs. nontumorigenic and dormant tumor. | [34] |
| VEGF(189) overexpression | Inducer of angiogenic dormancy | ||
| Thrombospondin-1 | Inducer of angiogenic dormancy | Endothelial-derived Thrombospondin-1 induces long-lasting BCC dormancy. This repressive nod is lost in sprouting neovasculature where active TGFβ1 and periostin act as tumor-promoting factors derived from endothelial tip cells. | [35] |
| TGFβ1, Periostin | Inhibitor of angiogenic dormancy | ||
| Thrombospondin, Angiomotin, Tropomyosin, TGF-β2, P4HA1, EphA5, H2BK, IGFBP-5 | Inducer of angiogenic dormancy | Dormant tumors undergo a stable genetic reprogramming during their switch to the fast-growing phenotype by downregulation of angiogenesis inhibitors such as Thrombospondin and decreased the sensitivity of angiogenic tumors to angiostatin along with upregulation of angiogenesis-related genes. | [36] |
| EGFR-1, IGF-IR, CD73, PI3K, ESM-1, PIK3CB, TIMP-3 | Inhibitor of angiogenic dormancy | ||
| MME1(NM23) | Inducer of angiogenic dormancy | NM23 inhibits EGF-induced cell migration. Increase the expression of metastasis-related genes TIMP-1, E-Cadherin and β-Catenin, reduce the expression of VEGR, CD44V6, and MMP-2 and reduce metastasis. | [37] |
| Kai-1 (CD82) | Inducer of angiogenic dormancy | Binding of tumor cell surface-expressed Kai1 with endothelial DARC inhibit tumor cell proliferation, induce senescence by modulating the expression of TBX2 and p21 and suppress metastasis. | [38] |
| BRMS1 | Inducer of angiogenic dormancy | BRMS1 inhibits angiogenesis through blocking NF/KB activity. It can also reduce metastatic potential but not tumorigenicity. | [39] |
| HSP27 | Inhibitor of angiogenic dormancy | Downregulation of HSP27 associated with reduced endothelial cell proliferation and decreased secretion of VEGF-A, VEGF-C, and induction of long-term dormancy. | [40] |
| CTL response, MHC class I, NK cells | Inducer of immunologic dormancy | An activate CTL response can maintain immune equilibrium with metastatic dormant cells. Immune dormancy arrest cancer cell growth and promotes angiogenic control. | [41] |
| B7-H1 and B7.1 | Inducer of immunologic dormancy | Dormant tumor cells up-regulate B7-H1 and B7.1 and resist CTL-mediated lysis. | [42] |
| Macrophage (MΦs) | Inhibitor/inducer of immunologic dormancy | By forming gap junctional interactions with CSCs, the M2 MΦs promote cycling quiescence and carboplatin resistance. M1 MΦ-derived exosomes activated NFкB to reverse quiescent BCCs to cycling cells in vivo. | [43] |
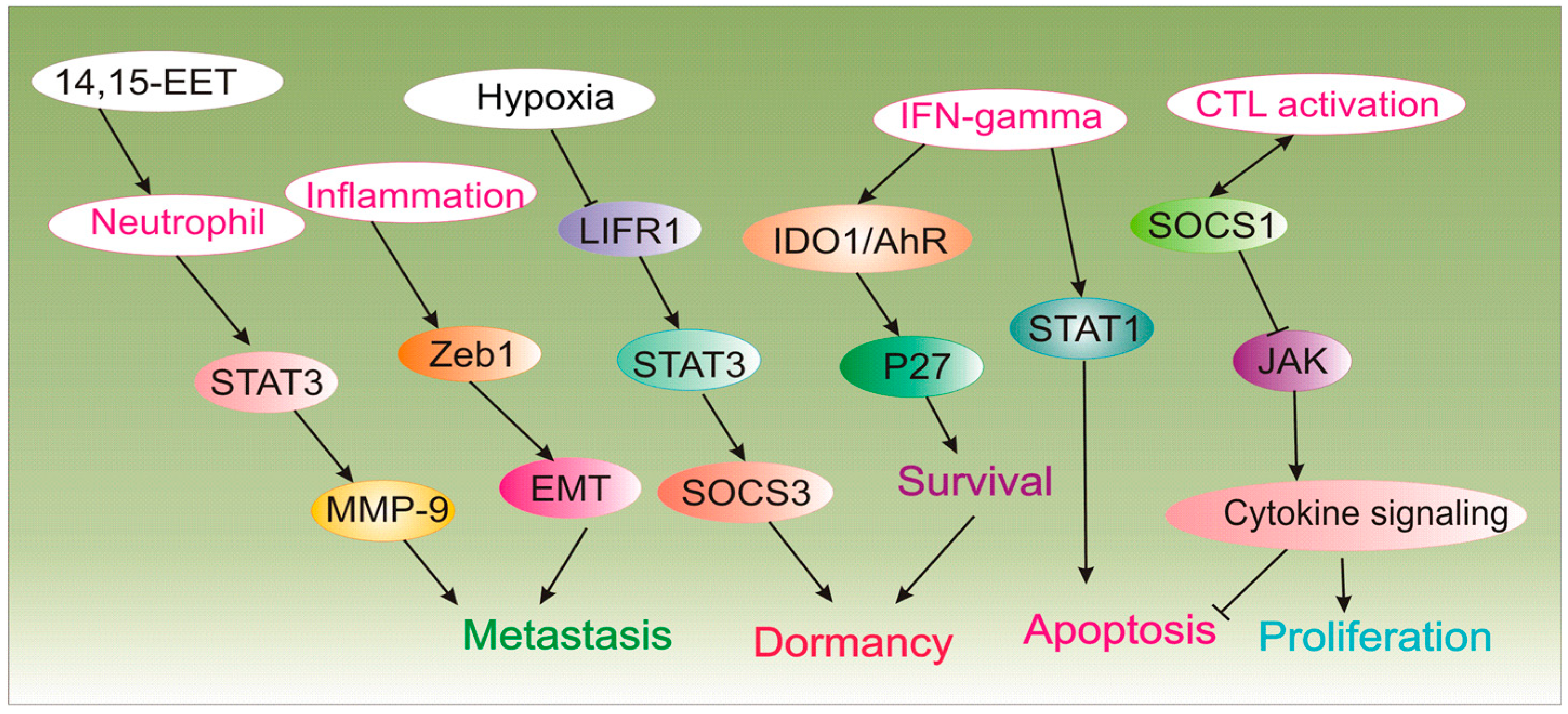
| Neutrophils | Inhibitor of immunologic dormancy | 14,15-EET trigger neutrophil infiltration in metastatic lesions by activating STAT3/JNK-hIL-8/mCXCL15 and mir-155 which converts tumor-suppressing function of neutrophils to tumor-promoting in vivo. In the presence of G-CSF/IL-6, 14,15-EET enhance STAT3 activation in neutrophils to decrease TRAIL expression and increase MMP-9 expression to induce angiogenesis during dormant micrometastases growth. Neutrophil depletion or blocking hIL-8/mCXCL15 abrogate micrometastases induced by 14,15-EET. | [44] |
| Zeb1 | Inhibitor of immunologic dormancy | Inflammation triggers Zeb1 to promote EMT and give rise to metastatic outgrowth. | [45] |
| TNFα, IL-β | Inhibitor of immunologic dormancy | Addition of bone remodeling cytokines, TNFα, and IL-β to dormant cancer cells induce proliferation and occurrence of latent bone metastasis. | [46] |
| SOCS1, IL-3 | Inducer of immunologic dormancy | T-cell inactivation and resistance to apoptosis are mediated by methylation of SOCS1, deregulation of JAK/STAT and overproduction of IL-3 by dormant cells. | [47] |
| IFN-γ | Inducer of immunologic dormancy | IFN-γ signaling triggers differentiated tumor cell apoptosis via STAT1; however, when IDO1 and AhR are overexpressed as in DTCs, IFN-gamma induces p27 via IDO1/AhR and inhibits STAT1 signaling, and favors dormancy state. | [48] |
| LPS/EGF | Inhibitor of immunologic dormancy | Activated immune/stromal cells stimulate the resident hepatic cells to derive tumor growth. | [9] |
| Mitochondrial dysfunction | Inhibitor of metabolic dormancy | VLX600 impairs OXPHOS and drives a HIF-1α-dependent switch to glycolysis, which this metabolic pathway can’t meet the energy demands of tumor cells, thus induction of autophagy is unavoidable. Yet, due to lack of HIF-1α-stabilization and glucose inaccessibility in metabolically stressed environments, shifting to glycolysis mode will be restricted, consequently, tumor cells undergo apoptosis. | [11] |
| LACTB | Inhibitor of metabolic dormancy | Mitochondrial tumor suppressor, LACTB potently inhibits the proliferation of BC cells via altering mitochondrial lipid metabolism and differentiation of BC cells by reduction of the levels of mitochondrial phosphatidylserine decarboxylase, which is involved in the synthesis of mitochondrial phosphatidylethanolamine. | [49] |
| FA metabolism, ROS, oxidative DNA damage | Inducer of metabolic dormancy | Residual cells display altered lipid metabolism, elevated ROS, and increased oxidative DNA damage. Thus, lipid metabolism and ROS are therapeutic targets for reducing tumor recurrence in BC patients. | [50] |
| NR2F1 | Inducer of hypoxic dormancy | Hypoxic HNSCC and breast primary tumor microenvironments display upregulation of key dormancy (NR2F1, DEC2, p27) and hypoxia (GLUT1, HIF1α) genes. Post-hypoxic DTCs were frequently NR2F1hi/DEC2hi/p27hi/TGFβ2hi, dormant and chemotherapy-resistant. | [51] |
| LIFR | Inducer of hypoxic dormancy | In BC patients with bone metastases, low LIFR levels negatively correlate with HIF-1α activity and disease outcome. Hypoxia reduces the LIFR: STAT3: SOCS3 signaling in BC cells. Loss of the LIFR or STAT3 reactivates dormant BC cells to proliferate and to downregulate stem cell-related genes and specifically benefit their bone colonization. | [52] |
| E6/E7 antigen | Inhibitor of hypoxic dormancy | Human papillomavirus-infected cancer cells can enter into reversible dormancy state, with reducing the synthesis of viral antigen and enhanced therapeutic resistance, and uphold tumor recurrence upon reoxygenation. | [53] |
| Kiss-1, CRSP3 | Inducer of ECM dormancy | Kiss-1 expression suppresses malignant melanoma metastasis, inhibits motility, chemotaxis, and invasion, perhaps by suppressing the expression of MMP-9. CRSP3 regulate the transcriptional expression of Kiss-1. | [54] |
| Type I collagen (Col-I) | Inhibitor of ECM dormancy | Atypical tetraspanin TM4SF1 as a potent inducer of metastatic recurrence of BC couples DDR1 to PKCα. This kinase activates JAK2. Then, JAK2/STAT3 activates the expression of SOX2 and NANOG, maintain the manifestation of CSC traits, and fuel metastatic recurrence in the bone, lung, and brain. | [55] |
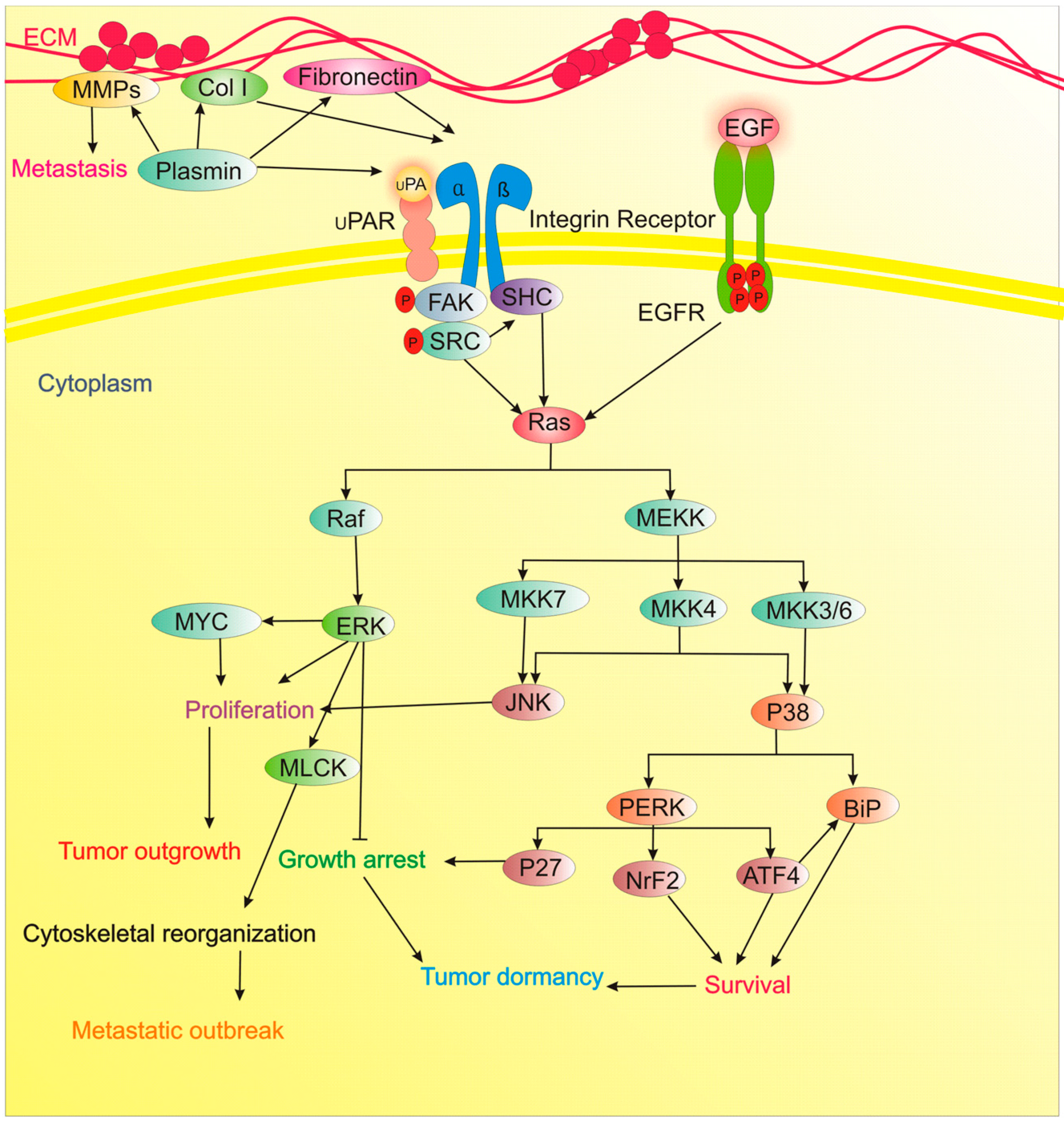
| Fibronectin | Inhibitor of ECM dormancy | Fibronectin/β1 Integrin/MLCK axis induces a transition from a quiescent to proliferative, metastatic outgrowth. | [56] |
| Col-I | Inhibitor of ECM dormancy | Col-I/β1 Integrin/SRC/FAK/ERK/MLCK signaling induce dormant cells to switch to proliferative metastatic lesions. | [57] |
| u-PAR | Inducer of ECM dormancy | u-PAR, is an essential molecule in BM disseminated tumor cells for long-standing survival during dormancy by regulation of u-PAR of α5β1 integrins, and signal propagation from Fibronectin through the p38, ERK, and EGF-receptor signaling. | [58] |
| FAK, Src, MEK1/2 (ERK1/2) | Inhibitor of ECM dormancy | Targeting Src prevents the proliferative response of dormant cells to external stimuli. MEK1/2 inhibition suppresses their survival and eliminates tumor relapse. | [59] |
| KRAS/C-Myc, IGF1/AKT | Inducer of ECM dormancy | KRAS/C-Myc negative dormant cells represent an increase in autocrine IGF1/AKT. Inhibition of IGF-1R reduces residual disease burden and cancer recurrence. | [60] |
| TGFB2 | Inducer of ECM dormancy | Cellular adhesion promotes PC cells to escape from dormancy and lethal metastasis. The mechanism involves downregulation of TGFB2, E2F4, and upregulation of MLCK, CDK6. | [61] |
| DIRAS family GTPase 3 (DIRAS3) | Inducer of ECM dormancy | DIRAS3 decrease ERK/AKT signaling and induce autophagy. Addition of VEGF, IGF-1, and IL-8 abrogate sustaining autophagic DIRAS3-induced dormancy. A combination of antibodies targeting VEGF, IGF-1, and IL-8 prevent outgrowth of dormant cells. | [62] |
| Aurora kinase A (AURKA) | Inhibitor of ECM dormancy | Activation of URKA-Erk1/2 signaling pathway induces chemotherapy resistance and promote metastasis of laryngeal squamous cell carcinoma. | [63] |
| Fbxw7 | Inducer of ECM dormancy | Dormant breast cancer cells overexpress Fbxw7, which acts as the negative control of cell cycle and its disruption avoids entry of dormant cells into the quiescent state, rendering them sensitive to chemotherapy. | [64] |
| Wnt5a | Inducer of ECM dormancy | Wnt5a/ROR2/SIAH2 signaling axis is involved in the induction and maintenance of PCa cells dormancy in the bone. | [65] |
| TGFβ2/ GDF10 | Inducer of ECM dormancy | Osteoblast-secreted proteins induce TGFβRIII-p38MAPK-pS249/T252RB pathway to mediate dormancy of metastatic PC in the bone. | [66] |
| Axl, Gas6 | Inducer of ECM dormancy | Axl is a tyrosine kinase receptor for growth arrest-specific 6 (Gas6). Axl and Gas6 are required for TGF-β2-induced dormancy of PC cells in the bone marrow. | [67] |
| E-selectin, SDF-1 | Inducer of ECM dormancy | Proliferating and dormant BCCs inhabit different regions, whereas E-selectin interactions allow BCC residency in the BM, the SDF-1/CXCR4 binding anchors BCCs to the metastatic niche. Blocking CXCR4 (SDF receptor) and E-selectin eliminates latent micrometastases residing in supportive bone, excising occurrence of relapsed disease. | [68] |
| MED12 | Inducer of ECM dormancy | The lack of MED12 induces tumor cell dormancy. Re-expression of MED12 abrogates tumor cell dormancy by positively controlling EGFR expression. | [69] |
| N-cadherin | Inducer of CSC dormancy | N-cadherin upregulation leads to downregulation of E-cadherin, upregulation of Connexin, EMT, and dormancy. | [70] |
| Notch | Inducer of CSC dormancy | Notch remain activated in dormant residual cells and accelerates tumor recurrence. | [71] |
| CD13 | Inducer of CSC dormancy | CD13 is a cancer stem cell dormancy marker in HCC. | [72] |
| Coco | Inhibitor of CSC dormancy | Coco enhances cancer stem cell traits and antagonizes TGF-β activity. Coco reactivates dormant BC cells in the lung whereas BMP signaling revives metastasis dormancy in the lung. | [73] |
| BMP7 | Inducer of CSC dormancy | Bone stromal cells-derived BMP7 stimulates senescence in prostate CSCs by activating BMP7-BMPR2/p38/p21/NDRG1 axis. | [74] |
| SPARC | Inducer of CSC dormancy | SPARC demethylation (activation) significantly stimulate the expression of BMP7 in bone marrow stromal cells and is required for BMP7 mediated stemness and senescence of PC cells. | [75] |
| SOX2 | Inducer of CSC dormancy | Low level of SOX2 expression is required for DTCs maintenance. SOX2 complete depletion of SOX results in activation of STAT3-p53-caspase axis and induction of cell apoptosis. | [76] |
| HMGA1 | Inhibitor of CSC dormancy | HMGA1 reprogram triple-negative BC cells to a stem-like state, driving their metastatic outgrowth. HMGA1 silencing excise cancer stem/initiator cells and prevents oncogenesis. | [77] |
| TBK1 | Inducer of CSC dormancy | PC cells target the HSC niche in mouse bone marrow during metastasis. Interaction with niche osteoblasts activate TBK1 expression and inhibit mTOR in PCa cells. Silencing TBK1 dampen drug resistance and formation of PCa stem-like cells. | [78] |
| p53, Necdin | Inducer of CSC Dormancy | Necdin-knock out adult HSCs is more proliferative and less quiescent than wild-type HSCs, indicating that Necdin resembles p53 function in supporting HSC dormancy during stable conditions. | [19] |
| PRRX1 | Inducer of CSC Dormancy | PRRX1 positively controls dormancy through TGF-β and promoting EMT in HNSCC and its activity is correlated with low expression of miR-642b-3p and TGF-β2 and p38. | [79] |
| Zeb1, G9a, SMAD5, SMARCD3, | Inhibitor of Epigenetic dormancy | These genes control EMT and control dormancy by reversible activation of stem cell-like properties of breast cancer cells in vitro. | [80] |
| KAT5, DOT1L | Inducer of epigenetic dormancy | These genes are involved in MET, promoting epithelial morphologies and thus reduce invasive properties of breast cancer cells in vitro. | |
| PCL1 | Inducer of epigenetic dormancy | PCL2 and PCL3 are expressed in proliferative tumor state, whereas PCL1 mainly expressed in dormant cells. | [81] |
| PCL2,3 | Inhibitor of epigenetic dormancy | ||
| NR2F1 | Inducer of epigenetic dormancy | NR2F1 is epigenetically upregulated in tumors and induce dormancy by global chromatin repression. | [82] |
| MSK1 | Inducer of epigenetic dormancy | MSK1 epigenetically controls the differentiation of cancer cells and its expression promotes metastatic dormancy. | [83] |
| miR-222/223 | Inducer of Dmir dormancy | Promotes quiescence and drug resistance. | [84] |
| miR-34a, miR-93, miR-200c | Inducer of Dmir dormancy | Loss of DmiRNAs happens during the transition from avascular dormant into angiogenic fast-growing phenotype. | [85] |
| Mir16/19, miR-580, 588 or 190 | Inducer of Dmir dormancy | Dmirs govern tumor dormancy, especially miR-190 induce long-lasting dormancy in glioblastomas and osteosarcomas. | [17] |
| miR-100-5p | Inducer of Dmir dormancy | miR-100-5p inhibition induces apoptosis in dormant PC cells and prevents the emergence of castration-resistant PC. | [86] |
| miR-200b/200a/429 | Inducer of Dmir dormancy | Expression of these Dmirs induce tumor cell dormancy and inhibit lung metastasis of BCCs. | [87] |
| miR-125b | Inducer of Dmir dormancy | Its expression favors epithelial phenotype, reduces Wnt-associated stem cell signaling and mesenchymal-associated genes and thus reduce metastasis of BCCs to the bone. | [88] |
| p38 Signaling | Inducer of stress-induced dormancy | p38 Up, ERK down leads to tumor dormancy. | [89] |
| Inhibitor of stress-induced dormancy | p38down/ERKup leads to mitogenesis. | ||
| Inducer of stress-induced dormancy | p38/BiP/PERK axis promotes drug resistance and survival of quiescent cells. BiP up-regulation averts Bax activation. | [90] | |
| Inducer of stress-induced dormancy | p38 induces dormancy by expression of p53 and BHLHB3 while inhibiting c-Jun and FoxM1. | [91] | |
| Inducer of stress-induced dormancy | MKK6 and p38α/β induce survival by regulating nuclear translocation and transcriptional activation of ATF6α in dormant cancer cells. | [92] | |
| Inducer of stress-induced dormancy | TGF-β2-MAPK p38α/β-(ERK/p38) (low)- DEC2/SHARP1, p27, ↓CDK4, and dormancy of malignant DTCs. | [93] | |
| Inducer of stress-induced dormancy | ˧MERTK, the ↓ratio of P-Erk1/2 to P-p38, ↑ p27, NR2F1, SOX2, and NANOG, ↑histone H3K9me3 and H3K27me3, G1/G0 arrest and dormancy. | [94] | |
| Inducer of stress-induced dormancy | MKK4 activates MAPK, p38 and JNK, up-regulate p21 and induce cancer cell growth arrest. | [95] | |
| Inducer of stress-induced dormancy | MKK6 activates MAPK and p38. | [95] | |
| Inducer of stress-induced dormancy | MKK7 activates MAPK and JNK. | [96] |
| Trial Name | Phase | ClinicalTrials.gov Identifier | Anti- Dormancy Strategy | Targeting Agent | End-Point | Results |
|---|---|---|---|---|---|---|
| Secondary adjuvant treatment for patients with isolated tumor cells in bone marrow | II | NCT00248703 | Addition of docetaxel in the adjuvant treatment to reduces the risk of persistent DTCs | docetaxel | Disease-free survival by DTC status; DTC number in BM aspirate | DTC eradication in 79% of patients; enhanced metastasis-free survival |
| CLEVER pilot trial: A phase II pilot trial of hydroxychloroquine, everolimus or the combination for prevention of recurrent breast cancer | II | NCT03032406 | Target persistent DTCs following standard of care treatment in breast cancer patients | hydroxychloroquine, everolimus or combination | Number of adverse events; DTC number in BM aspirate | N/A |
| Zoledronic acid in the treatment of breast cancer with minimal residual disease in the bone marrow (MRD-1) | II | NCT00172068 | Inhibitor of bone resorption; interrupt dormancy state of DTCs | zoledronic acid + calcium/vitamin D | Reduction of detected tumor cells in BM | N/A |
| A phase Ib/II trial of gedatolisib, hydroxychloroquine or the combination of prevention of recurrent breast cancer (GLACIER) | Ib/II | CT03400254 | Target persistent DTCs following standard of care treatment in breast cancer patients | hydroxychloroquine, gedatolisib or combination | DTC number in BM aspirate | N/A |
| A pilot study to evaluate the impact of denosumab on disseminated tumor cells in patients with early-stage breast cancer | II | NCT01545648 | Interrupt immunological dormancy by blocking RNKL overexpression by DTCs which foster the production of chronic inflammatory cytokines. | denosumab | DTC number in BM aspirate | N/A |
| A pilot study of mobilization and treatment of disseminated tumor cells in men with metastatic prostate cancer | I | NCT02478125 | Anti-CXCR4 strategy can be used to mobilize and target persistent DTCs | burixafor hydrobromide, G-CSF, docetaxel, or combination | CTC number in peripheral blood; HSC number in peripheral blood; PSA response, safety | N/A |
| A pilot study of the combination of 5-azacitidine (5-AZA) and all-trans retinoic acid (ATRA) for prostate cancer (PCa) with PSA only recurrence after definitive local treatment | II | NCT03572387 | Reprogramming therapy in patients with recurrent PCa based on rising PSA only. | Combination of 5-azacitidine and alltrans retinoic acid, and lupron | Disease progression-free rate, Percentage of adverse events by grade, time to tumor progression, measurement of dormancy markers TGF-β2, BMP7, BMP4, GAS6, retinoic acid and NR2F1 | N/A |
| Effect of trastuzumab on disease-free survival in early-stage HER2-negative breast cancer patients with ERBB2 expressing disseminated tumor cells | II | NCT01779050 | Targeted trastuzumab therapy to eliminate HER2 expressing disseminated tumor cells in BM. | doxorubicin, trastuzumab, cyclophosphamide, paclitaxel, epirubicin, docetaxel, carboplatin, fluorouracil, and combination | Elimination of ERBB2-positive DTCs from BM. Improved disease-free survival. | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahanban-Esfahlan, R.; Seidi, K.; Manjili, M.H.; Jahanban-Esfahlan, A.; Javaheri, T.; Zare, P. Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer. Cancers 2019, 11, 1207. https://doi.org/10.3390/cancers11081207
Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P. Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer. Cancers. 2019; 11(8):1207. https://doi.org/10.3390/cancers11081207
Chicago/Turabian StyleJahanban-Esfahlan, Rana, Khaled Seidi, Masoud H. Manjili, Ali Jahanban-Esfahlan, Tahereh Javaheri, and Peyman Zare. 2019. "Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer" Cancers 11, no. 8: 1207. https://doi.org/10.3390/cancers11081207
APA StyleJahanban-Esfahlan, R., Seidi, K., Manjili, M. H., Jahanban-Esfahlan, A., Javaheri, T., & Zare, P. (2019). Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer. Cancers, 11(8), 1207. https://doi.org/10.3390/cancers11081207