Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
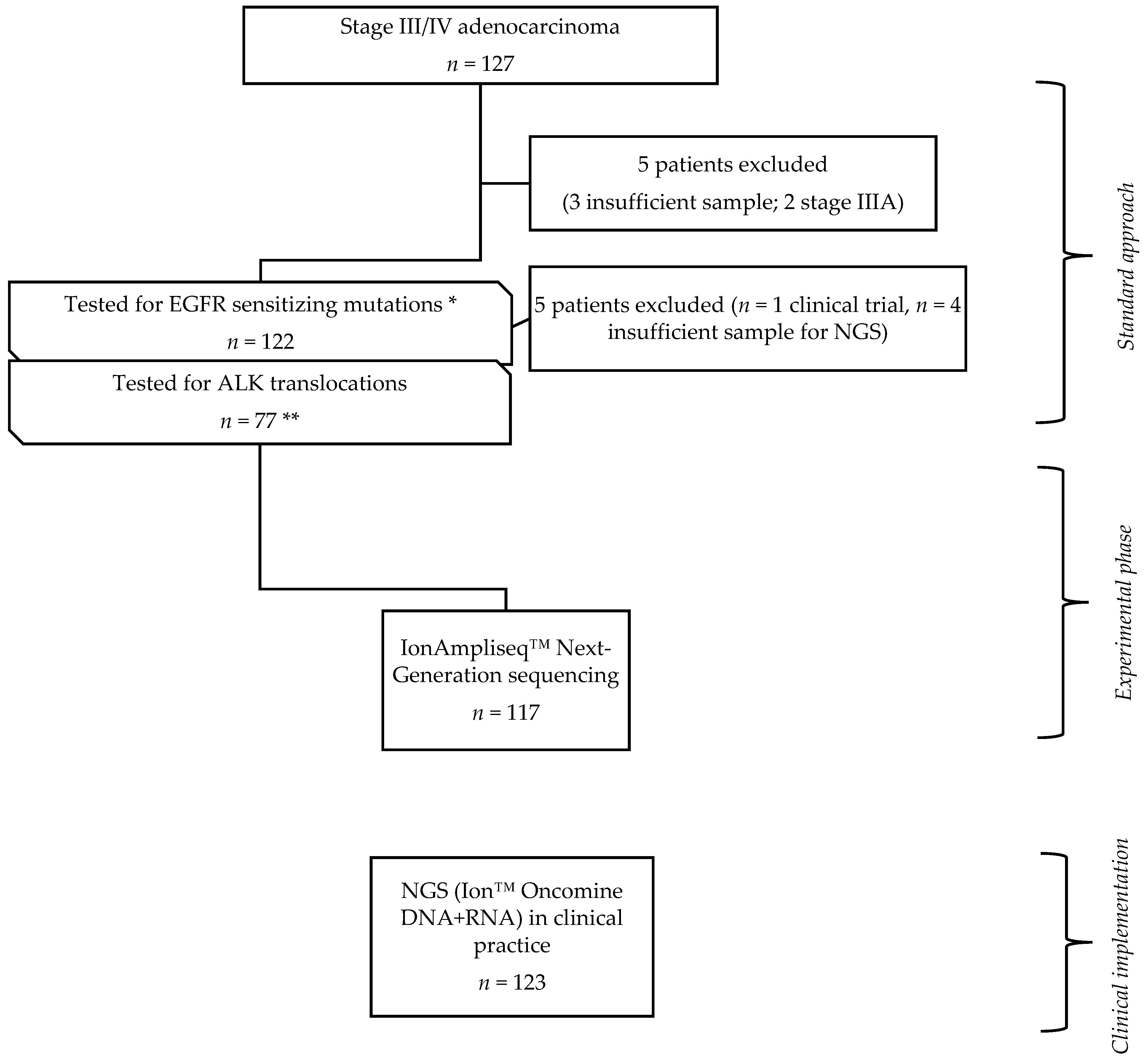
2.1. Study Design
2.2. Tumor Specimens
2.3. Library and Template Preparation for Next-Generation Sequencing
2.4. Next-Generation Sequencing and Bioinformatic Analysis
2.5. Statistical Analysis
3. Results
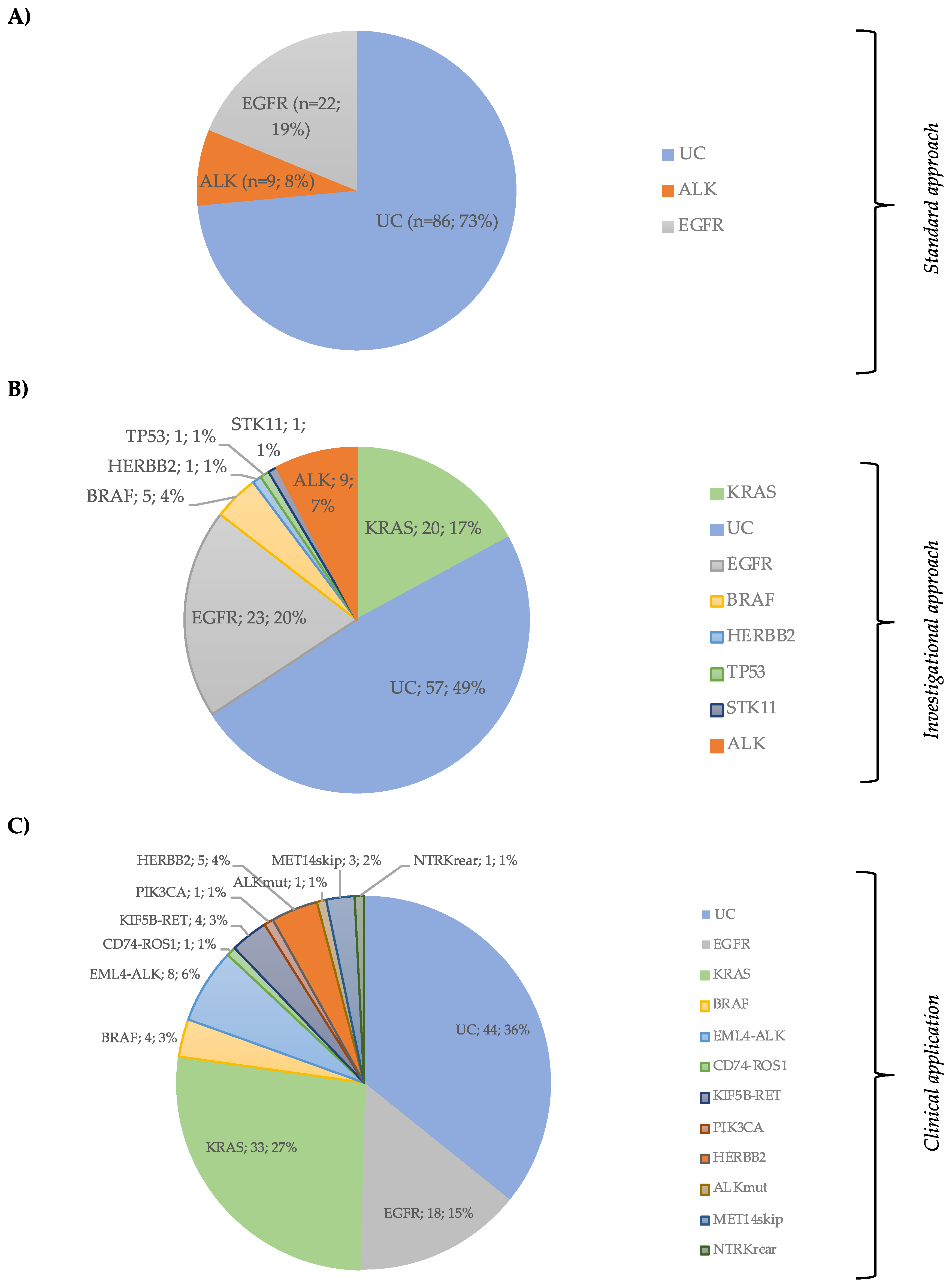
3.1. Clinicopathologic Characteristics and Mutational Profile
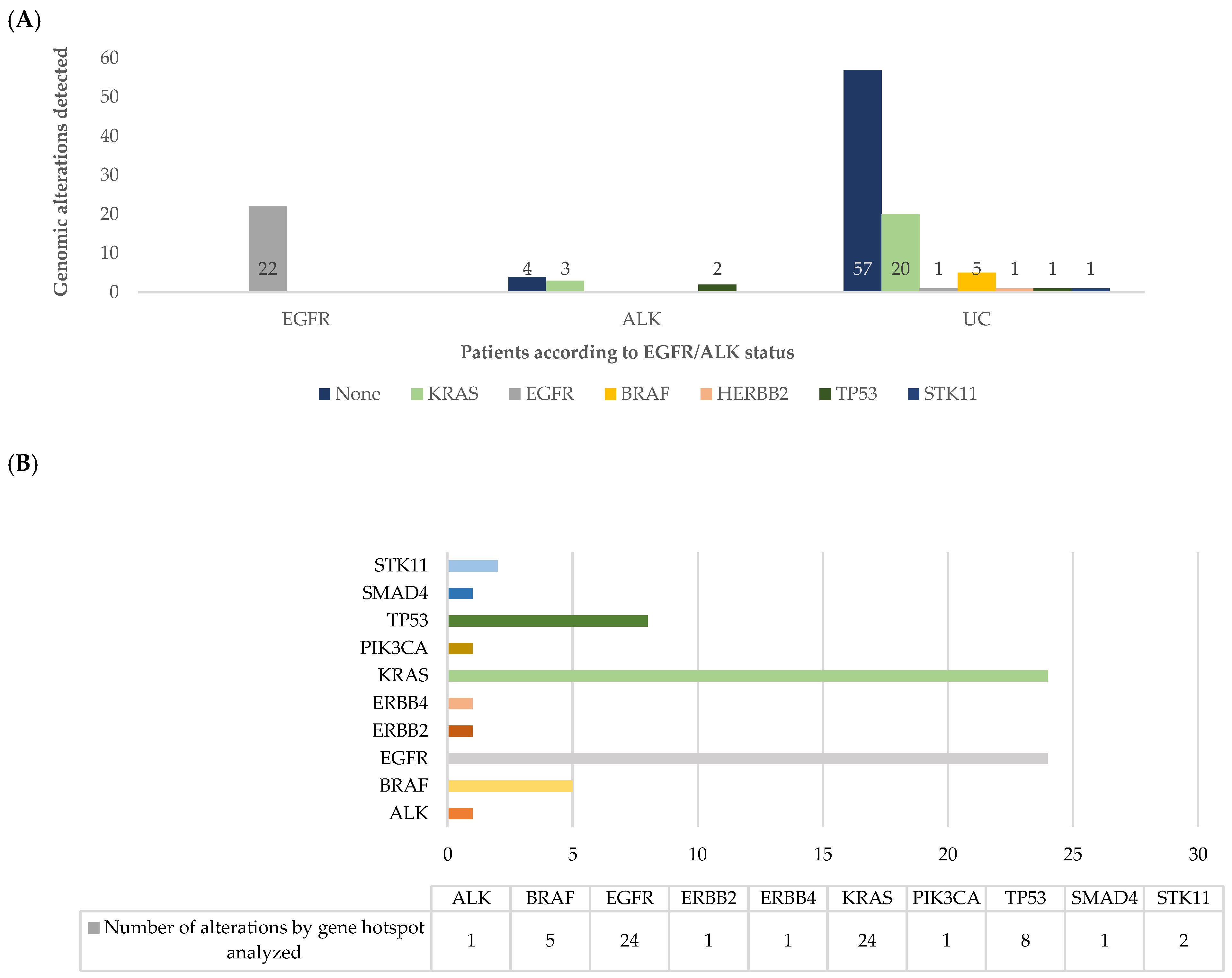
3.2. Next-Generation Sequencing Results
3.3. Concurrent Genomic Alterations
3.4. Concordance Between Sanger and NGS for the EGFR Status
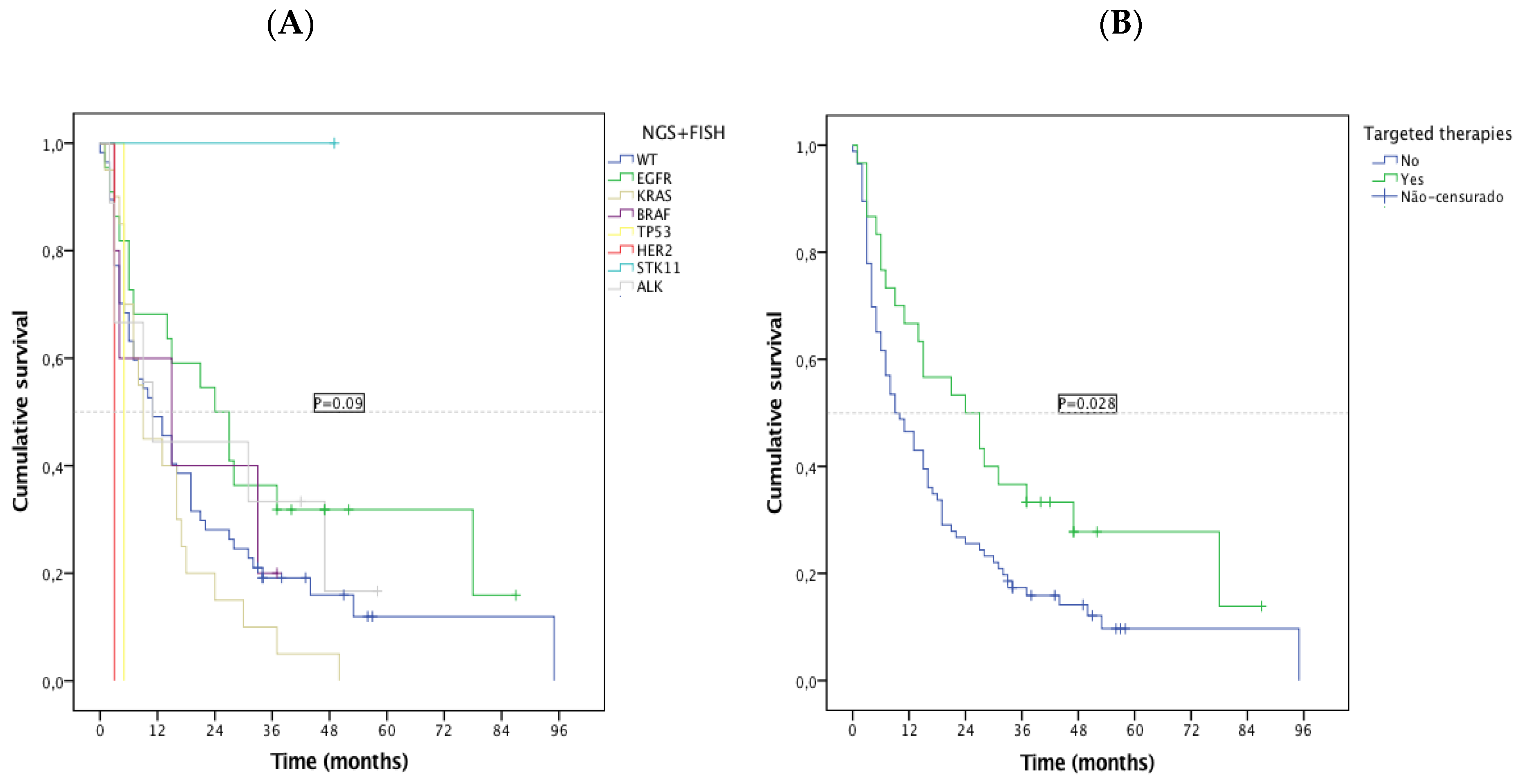
3.5. Impact of the Identification of Targetable Alterations in Patient’s Overall Survival
3.6. NGS Results from the Implementation Cohort with a Combined DNA and RNA Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Shames, D.S.; Wistuba, I.I. The Evolving Genomic Classification of Lung Cancer. J. Pathol. 2014, 232, 121–133. [Google Scholar] [CrossRef]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of Four Chemotherapy Regimens for Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Thorac. Oncol. 2013, 8, 823–859. [Google Scholar]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Riely, G.L. What, When, and How of Biomarker Testing in Non-Small Cell Lung Cancer. J. Natl. Compr. Canc. Netw. 2017, 15, 686–688. [Google Scholar] [CrossRef]
- Lim, C.; Tsao, M.S.; Le, L.W.; Shepherd, F.A.; Feld, R.; Burkes, R.L.; Liu, G.; Kamel-Reid, S.; Hwang, D.; Tanguay, J.; et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall-cell lung cancer. Ann. Oncol. 2015, 26, 1415–1421. [Google Scholar] [CrossRef]
- Gutierrez, M.E.; Choi, K.; Laan, R.B.; Licitra, E.J.; Skrzypczak, S.M.; Pe Benito, R.; Wu, T.; Arunajadai, S.; Kaur, S.; Harper, H.; et al. Genomic Profiling of Advanced Non-Small Cell Lung Cancer in Community Settings: Gaps and Opportunities. Clin. Lung Cancer 2017, 18, 651–659. [Google Scholar] [CrossRef]
- Groome, P.A.; Bolejack, V.; Crowley, J.J.; Kennedy, C.; Krasnik, M.; Sobin, L.H.; Goldstraw, P. The IASLC Lung Cancer Staging Project: Validation of the proposals for revision of the T, N, and M descriptors and consequent stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J. Thorac. Oncol. 2007, 2, 694–705. [Google Scholar] [CrossRef]
- Detterbeck, F.C.; Boffa, D.J.; Kim, A.W.; Tanoue, L.T. The Eighth Edition Lung Cancer Stage Classification. Chest 2017, 151, 193–203. [Google Scholar] [CrossRef]
- Treece, A.L.; Montgomery, N.D.; Patel, N.M.; Civalier, C.J.; Dodd, L.G.; Gulley, M.L.; Booker, J.K.; Weck, K.E. FNA smears as a potential source of DNA for targeted next-generation sequencing of lung adenocarcinomas. Cancer Cytopathol. 2016, 124, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Roy-Chowdhuri, S.; Goswami, R.S.; Chen, H.; Patel, K.P.; Routbort, M.J.; Singh, R.R.; Broaddus, R.R.; Barkoh, B.A.; Manekia, J.; Yao, H.; et al. Factors affecting the success of next-generation sequencing in cytology specimens. Cancer Cytopathol. 2015, 123, 659–668. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. JMD 2015, 17, 251–264. [Google Scholar]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef]
- D’Haene, N.; Le Mercier, M.; De Nève, N.; Blanchard, O.; Delaunoy, M.; El Housni, H.; Dessars, B.; Heimann, P.; Remmelink, M.; Demetter, P.; et al. Clinical Validation of Targeted Next Generation Sequencing for Colon and Lung Cancers. PLoS ONE 2015, 10, e0138245. [Google Scholar] [CrossRef]
- Tops, B.B.; Normanno, N.; Kurth, H.; Amato, E.; Mafficini, A.; Rieber, N.; Le Corre, D.; Rachiglio, A.M.; Reiman, A.; Sheils, O.; et al. Development of a semi-conductor sequencing-based panel for genotyping of colon and lung cancer by the Onconetwork consortium. BMC Cancer 2015, 15, 26. [Google Scholar] [CrossRef]
- Scarpa, A.; Sikora, K.; Fassan, M.; Rachiglio, A.M.; Cappellesso, R.; Antonello, D.; Amato, E.; Mafficini, A.; Lambiase, M.; Esposito, C.; et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS ONE 2013, 8, e80478. [Google Scholar] [CrossRef]
- Qiu, T.; Guo, H.; Zhao, H.; Wang, L.; Zhang, Z. Next-generation sequencing for molecular diagnosis of lung adenocarcinoma specimens obtained by fine needle aspiration cytology. Scientific Reports 2015, 5, 11317. [Google Scholar] [CrossRef] [Green Version]
- Kanagal-Shamanna, R.; Portier, B.P.; Singh, R.R.; Routbort, M.J.; Aldape, K.D.; Handal, B.A.; Rahimi, H.; Reddy, N.G.; Barkoh, B.A.; Mishra, B.M.; et al. Next-generation sequencing-based multi-gene mutation profiling of solid tumors using fine needle aspiration samples: Promises and challenges for routine clinical diagnostics. Mod. Pathol. 2014, 27, 314–327. [Google Scholar] [CrossRef]
- Karnes, H.E.; Duncavage, E.J.; Bernadt, C.T. Targeted next-generation sequencing using fine-needle aspirates from adenocarcinomas of the lung. Cancer Cytopathol. 2014, 122, 104–113. [Google Scholar] [CrossRef]
- McCourt, C.M.; McArt, D.G.; Mills, K.; Catherwood, M.A.; Maxwell, P.; Waugh, D.J.; Hamilton, P.; O’Sullivan, J.M.; Salto-Tellez, M.; et al. Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS ONE 2013, 8, e69604. [Google Scholar] [CrossRef]
- Tuononen, K.; Maki-Nevala, S.; Sarhadi, V.K.; Wirtanen, A.; Ronty, M.; Salmenkivi, K.; Andrews, J.M.; Telaranta-Keerie, A.I.; Hannula, S.; Lagstrom, S.; et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Gene. Chromosome. Cancer 2013, 52, 503–511. [Google Scholar]
- Marchetti, A.; Del Grammastro, M.; Filice, G.; Felicioni, L.; Rossi, G.; Graziano, P.; Sartori, G.; Leone, A.; Malatesta, S.; Iacono, M.; et al. Complex Mutations & Subpopulations of Deletions at Exon 19 of EGFR in NSCLC Revealed by Next Generation Sequencing: Potential Clinical Implications. PLoS ONE 2012, 7, e42164. [Google Scholar]
- Aguiar, F.; Fernandes, G.; Queiroga, H.; Machado, J.C.; Cirnes, L.; Souto Moura, C.; Hespanhol, V. Overall Survival Analysis and Characterization of an EGFR Mutated Non-Small Cell Lung Cancer (NSCLC) Population. Arch. Bronconeumol. 2018, 54, 10–17. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, E.Y.; Kim, H.R.; Ali, S.M.; Greenbowe, J.R.; Shim, H.S.; Chang, H.; Lim, S.; Paik, S.; Cho, B.C. Genomic profiling of lung adenocarcinoma patients reveals therapeutic targets and confers clinical benefit when standard molecular testing is negative. Oncotarget. 2016, 7, 24172–24178. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Pan, W.; Yang, Y.; Zhu, H.; Zhang, Y.; Zhou, R.; Sun, X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: A meta-analysis of 41 studies. Oncotarget 2016, 7, 8373–8388. [Google Scholar] [CrossRef]
- Schmid, S.; Gautschi, O.; Rothschild, S.; Mark, M.; Froesch, P.; Klingbiel, D.; Reichegger, H.; Jochum, W.; Diebold, J.; Früh, M. Clinical Outcome of ALK Positive Non-Small Cell Lung Cancer (NSCLC) Patients with De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs). J. Thorac. Oncol. 2017, 12, 681–688. [Google Scholar] [CrossRef]
- De Marchi, F.; Haley, L.; Fryer, H.; Ibrahim, J.; Beierl, K.; Zheng, G.; Gocke, C.D.; Eshleman, J.R.; Belchis, D.; Illei, P.; et al. Clinical Validation of Coexisting Activating Mutations Within EGFR, Mitogen-Activated Protein Kinase, and Phosphatidylinositol 3-Kinase Pathways in Lung Cancers. Arch. Pathol. Lab. Med. 2019, 143, 174–182. [Google Scholar] [CrossRef]
- Williams, H.L.; Walsh, K.; Diamond, A.; Oniscu, A.; Deans, Z.C. Validation of the Oncomine™ focus panel for next-generation sequencing of clinical tumour samples. Virchows Arch. 2018, 473, 489–503. [Google Scholar] [CrossRef]
- Suh, J.H.; Johnson, A.; Albacker, L.; Wang, K.; Chmielecki, J.; Frampton, G.; Gay, L.; Elvin, J.A.; Vergilio, J.A.; Ali, S.; et al. Comprehensive Genomic Profiling Facilitates Implementation of the National Comprehensive Cancer Network Guidelines for Lung Cancer Biomarker Testing and Identifies Patients Who May Benefit From Enrollment in Mechanism-Driven Clinical Trials. Oncologist 2016, 21, 684–691. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | EC Value (n, %) | IC Value (n, %) | p Value | |||
|---|---|---|---|---|---|---|
| Age (median, range) | 66 (38, 92) | 67 (41, 94) | 0.320 | |||
| Gender | Male | 71 (60.7) | 77 (62.6) | 0.760 | ||
| Female | 46 (39.3) | 46 (37.4) | ||||
| Performance status | 0 | 42 (35.9) | 61 (49.6) | 0.055 | ||
| 1 | 53 (45.3) | 38 (30.9) | ||||
| 2 | 16 (13.7) | 13 (10.6) | ||||
| 3 | 6 (5.1) | 11 (8.9) | ||||
| Smoking status | Smoker/Former Smoker | 74 (63.2) | 82 (66.7) | 0.579 | ||
| Never smoker | 43 (36.8) | 41 (33.3) | ||||
| Disease stage | III (A/B/C) | 22 (18.8) | 23 (18.7) | 0.984 | ||
| IV | 95 (81.2) | 100 (81.3) | ||||
| Histology | Adenocarcinoma | 116 (99.1) | 123 (100) | 0.304 | ||
| Adenosquamous | 1 (0.9) | 0 | ||||
| TTF1 IHQ | Positive | 101 (86.3) | 100 (81.3) | 0.070 | ||
| Negative | 5 (4.3) | 15 (12.2) | ||||
| Unknown | 11 (9.4) | 8 (6.5) | ||||
| Specimen type | Histologic | Bronchial | 31 (26.5) | 22 (17.8) | 0.487 | |
| Lung | 51 (43.6) | 63 (51.2) | ||||
| Pleura | 10 (8.5) | 7 (5.7) | ||||
| Brain | 2 (1.7) | 5 (4.1) | ||||
| Bone | 2 (1.7) | 1 (0.8) | ||||
| Liver | 0 | 2 (1.6) | ||||
| Lymph node | 0 | 3 (2.4) | ||||
| Skin | 0 | 1 (0.8) | ||||
| Small bowel | 0 | 1 (0.8) | ||||
| Total | 96 (82.1) | 105 (85.4) | ||||
| Cytologic | Lung-FNA | 2 (1.7) | 0 | |||
| EBUS-FNA | 2 (1.7) | 2 (1.6) | ||||
| Pleural fluid | 12 (10.2) | 10 (8.1) | ||||
| Pericardial fluid | 1 (0.9) | 0 | ||||
| Bronchial washing or brushing | 0 | 4 (3.2) | ||||
| Lymph node | 4 (3.4) | 2 (1.6) | ||||
| Total | 21 (17.9) | 18 (14.6) | ||||
| Molecular status (Standard approach) | EGFR | Mutated | 22 (18.8) | |||
| ALK | EML4-ALK | 9 (7.7) | ||||
| UC | 86 (73.5) | |||||
| Total | 117 (100) | |||||
| Patient’s Classification (Standard Approach) | NGS Genomic Alteration | AF | Clinical Significance | PFS1 | OS |
|---|---|---|---|---|---|
| UC | KRAS c.35G > T | 26.7 | Pathogenic | 7.1 | 9 |
| TP53 c.839G > A | 13.0 | Likely pathogenic | |||
| UC | BRAF c.1799T > A | 50.4 | Pathogenic | 4 | 15 |
| TP53 c.476C > G | 39.2 | Uncertain | |||
| UC | KRAS c.182A > G | 8.8 | Pathogenic | 2 | 5 |
| TP53 c.461G > T | 24.7 | Uncertain | |||
| STK11p.Glu199Asp | 12.8 | Pathogenic | |||
| UC | ERBB2 c.2310_2311insGCATAC | 20 | Not found | 0 | 3 |
| TP53 c.1024C > T | 21 | Pathogenic | |||
| UC | KRAS c.38_39delGCinsAA | 14.2 | n/a | 0 | 4 |
| ERBB4 c.1033G > T | 14.9 | n/a | |||
| EGFR | EGFR c.2235_2249del15 | 34.6 | Pathogenic | 10.1 | 37 (NR) |
| PIK3CA c.1633G > A | 27.8 | Pathogenic | |||
| EGFR | EGFR c.2236_2250del15 | 11.1 | Pathogenic | 8 | 24 |
| KRAS c.182A > G | 0.38 | Pathogenic | |||
| EGFR | EGFR c.2240_2257del18 | 67.2 | Pathogenic | 9.4 | 40 (NR) |
| EGFR c.2369C > T | 0.6 | Pathogenic | |||
| EGFR | EGFR c.2239_2248del | 66 | Pathogenic | 16.6 | NR |
| ALK c.3512T > A | 0.08 | Pathogenic | |||
| EML4-ALK | TP53 c.538G > T | 37.8 | Pathogenic | 9.7 | 42 |
| EML4-ALK | TP53 c.524G > A | 2.13 | Pathogenic | 16 | 31 |
| EML4-ALK | KRAS c.35G > T | 27.4 | Pathogenic | NE | 3 |
| EML4-ALK | KRAS c.35G > T | 11.7 | Pathogenic | 7 | 47 |
| EML4-ALK | KRAS c.35G > A | 6.1 | Pathogenic | NE | 2 |
| Gene | Cases Compared (n) | Concordant Results SS vs. NGS | Discordant SS vs. NGS | Concordant Cases (%) | Kappa | ||
|---|---|---|---|---|---|---|---|
| Neg/Neg | Pos/Pos | Neg/Pos | Pos/Neg | ||||
| EGFR | 117 | 94 | 22 | 1 | 0 | 99.1 | 0.972 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, M.G.O.; Jacob, M.; Martins, N.; Moura, C.S.; Guimarães, S.; Reis, J.P.; Justino, A.; Pina, M.J.; Cirnes, L.; Sousa, C.; et al. Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation. Cancers 2019, 11, 1229. https://doi.org/10.3390/cancers11091229
Fernandes MGO, Jacob M, Martins N, Moura CS, Guimarães S, Reis JP, Justino A, Pina MJ, Cirnes L, Sousa C, et al. Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation. Cancers. 2019; 11(9):1229. https://doi.org/10.3390/cancers11091229
Chicago/Turabian StyleFernandes, Maria Gabriela O., Maria Jacob, Natália Martins, Conceição Souto Moura, Susana Guimarães, Joana Pereira Reis, Ana Justino, Maria João Pina, Luís Cirnes, Catarina Sousa, and et al. 2019. "Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation" Cancers 11, no. 9: 1229. https://doi.org/10.3390/cancers11091229
APA StyleFernandes, M. G. O., Jacob, M., Martins, N., Moura, C. S., Guimarães, S., Reis, J. P., Justino, A., Pina, M. J., Cirnes, L., Sousa, C., Pinto, J., Marques, J. A., Machado, J. C., Hespanhol, V., & Costa, J. L. (2019). Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation. Cancers, 11(9), 1229. https://doi.org/10.3390/cancers11091229