Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. General Parameters
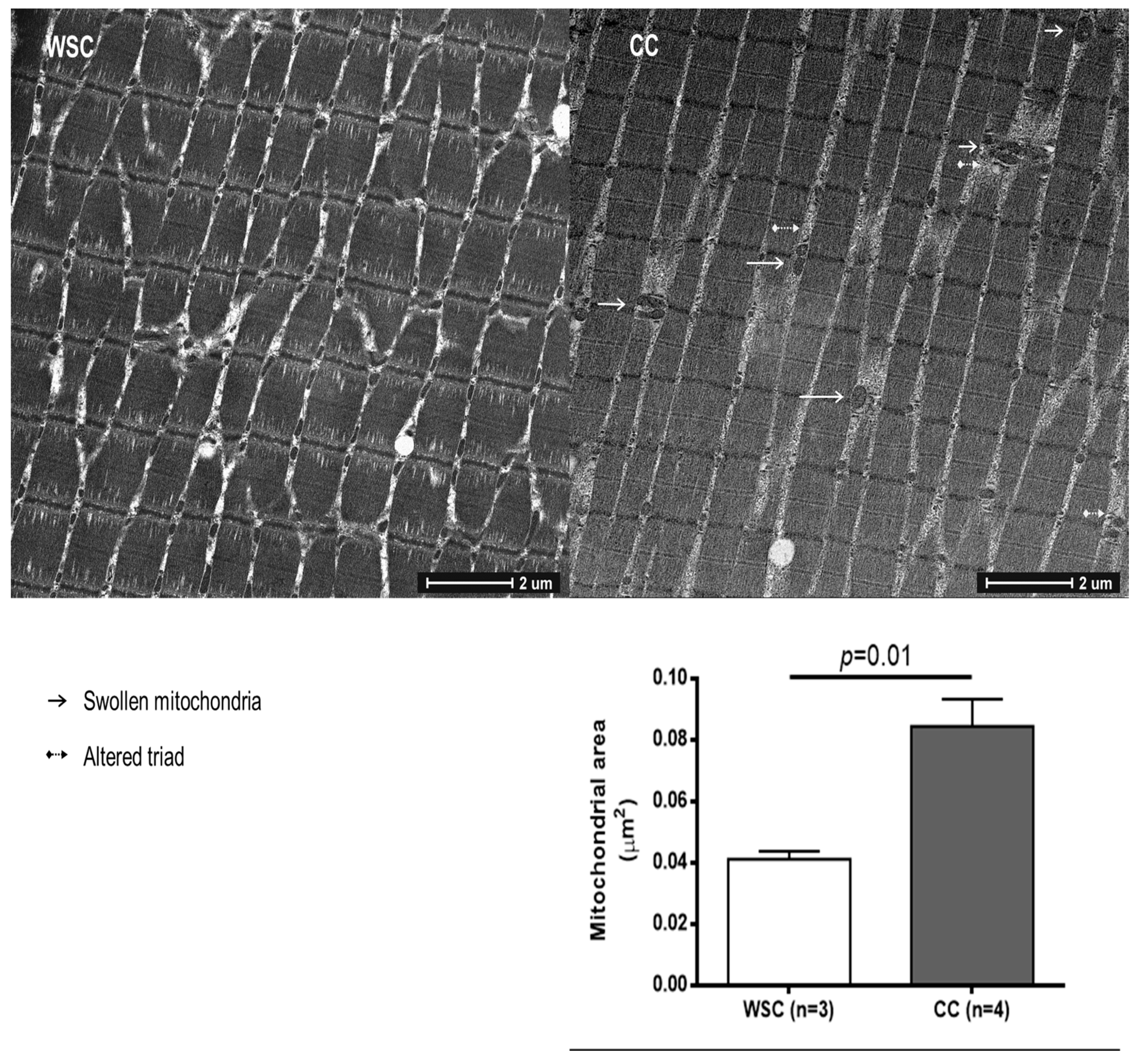
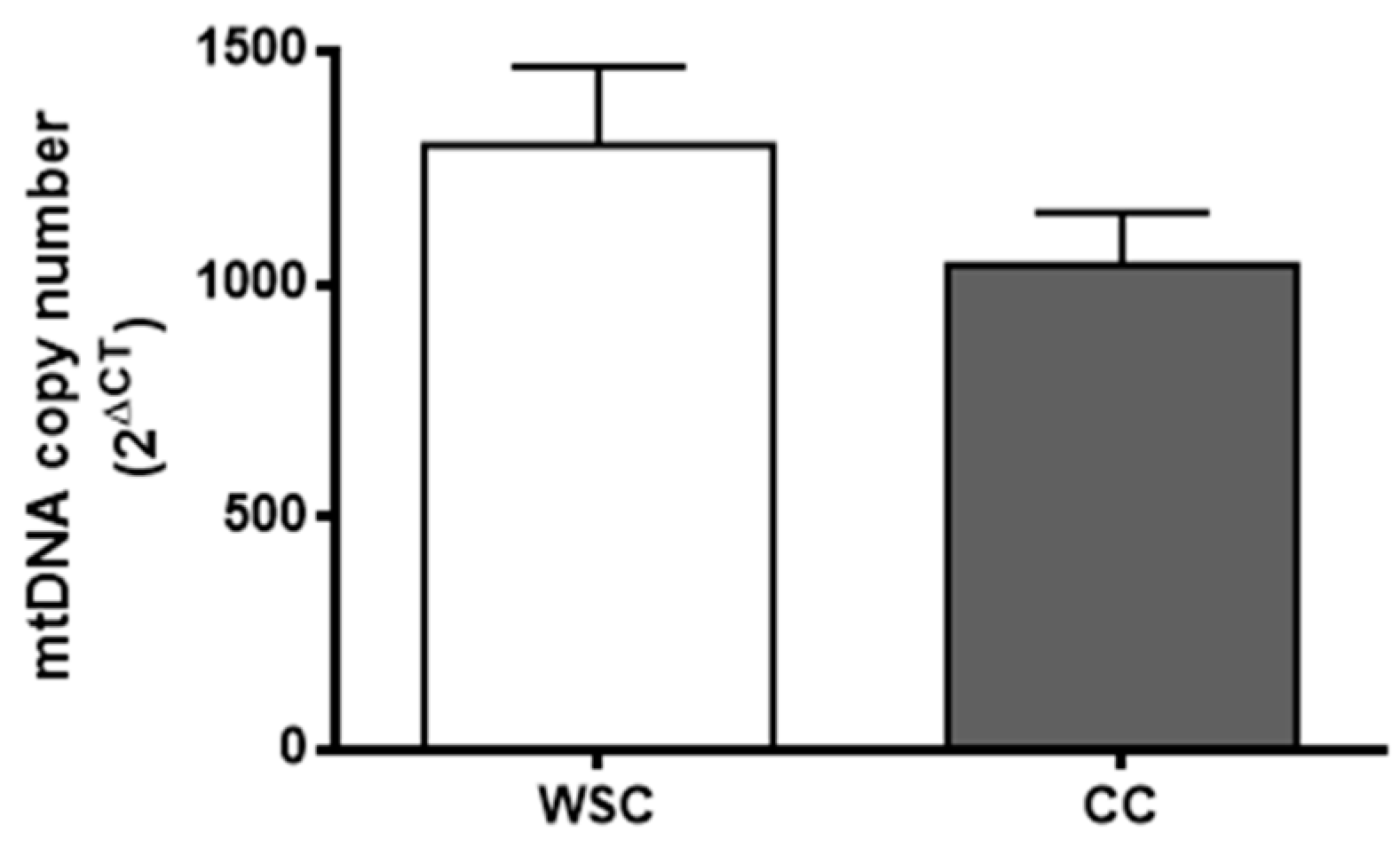
2.2. Mitochondrial Alterations
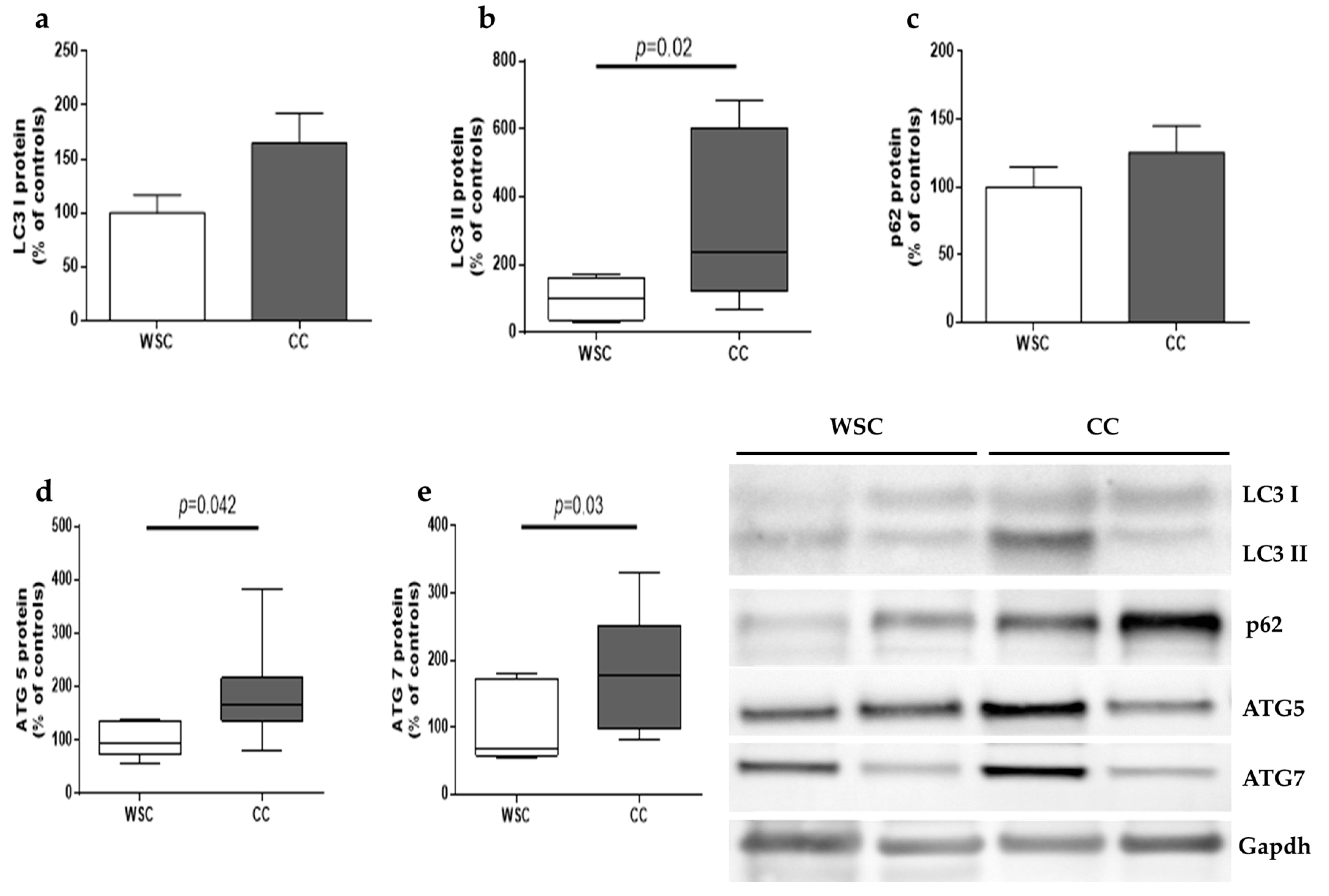
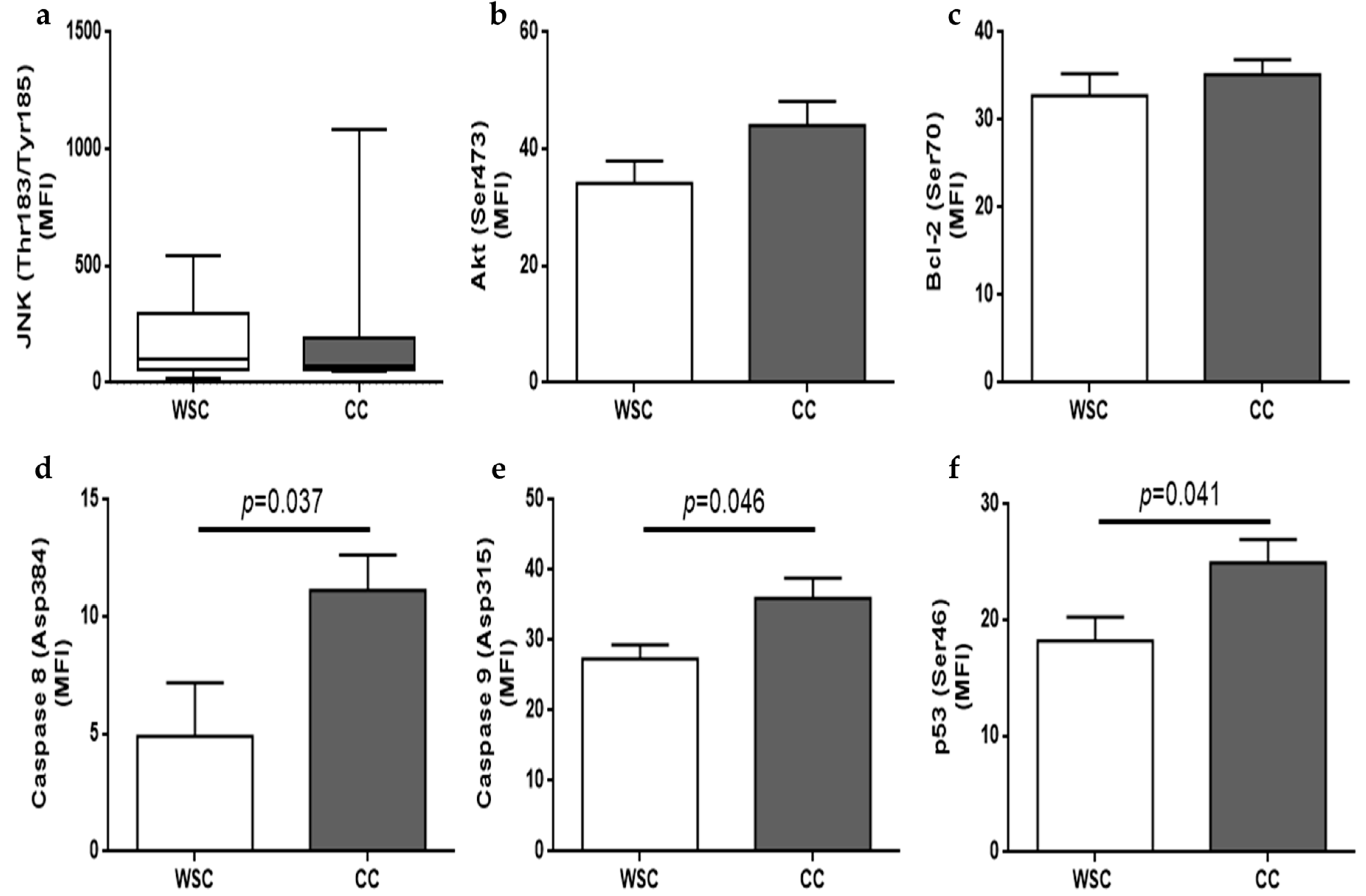
2.3. Autophagy and Apoptosis
3. Discussion
4. Materials and Methods
4.1. Patients Recruitment
4.2. Blood and Serum Analyses
4.3. Transmission Electron Microscopy and Mitochondrial Area
4.4. Gene Expression
4.5. Skeletal Muscle Protein Quantification and Western Blot Analysis
4.6. Apoptosis-Related Proteins Quantification
4.7. Mitochondrial DNA Copy Number
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Evans, W.J.; Morley, J.E.; Argilés, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef]
- Neves, R.X.; Rosa-Neto, J.C.; Yamashita, A.S.; Matos-Neto, E.M.; Riccardi, D.M.R.; Lira, F.S.; Batista, M.L.; Seelaender, M. White adipose tissue cells and the progression of cachexia: Inflammatory pathways. J. Cachexia Sarcopenia Muscle 2016, 7, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Silvério, R.; Lira, F.S.; Oyama, L.M.; Oller do Nascimento, C.M.; Otoch, J.P.; Alcântara, P.S.M.; Batista, M.L.; Seelaender, M. Lipases and lipid droplet-associated protein expression in subcutaneous white adipose tissue of cachectic patients with cancer. Lipids Health Dis. 2017, 16, 159. [Google Scholar] [CrossRef] [PubMed]
- Batista, M.L.; Henriques, F.S.; Neves, R.X.; Olivan, M.R.; Matos-Neto, E.M.; Alcântara, P.S.M.; Maximiano, L.F.; Otoch, J.P.; Alves, M.J.; Seelaender, M. Cachexia-associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J. Cachexia Sarcopenia Muscle 2016, 7, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.J.; Figuerêdo, R.G.; Azevedo, F.F.; Cavallaro, D.A.; Neto, N.I.P.; Lima, J.D.C.; Matos-Neto, E.; Radloff, K.; Riccardi, D.M.; Camargo, R.G.; et al. Adipose tissue fibrosis in human cancer cachexia: The role of TGFβ pathway. BMC Cancer 2017, 8, 237–282. [Google Scholar] [CrossRef] [PubMed]
- De Matos-Neto, E.M.; Lima, J.D.; de Pereira, W.O.; Figuerêdo, R.G.; Riccardi, D.M.; Radloff, K.; das Neves, R.X.; Camargo, R.G.; Maximiano, L.F.; Tokeshi, F.; et al. Systemic inflammation in cachexia-Is tumor cytokine expression profile the culprit? Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Muscaritoli, M.; Bossola, M.; Aversa, Z.; Bellantone, R.; Rossi Fanelli, F. Prevention and treatment of cancer cachexia: New insights into an old problem. Eur. J. Cancer 2006, 42, 31–41. [Google Scholar] [CrossRef]
- Fontes-Oliveira, C.C.; Busquets, S.; Toledo, M.; Penna, F.; Aylwin, M.P.; Sirisi, S.; Silva, A.P.; Orpí, M.; García, A.; Sette, A.; et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 2770–2778. [Google Scholar] [CrossRef]
- Julienne, C.M.; Dumas, J.F.; Goupille, C.; Pinault, M.; Collin, A.; Tesseraud, S.; Couet, C.; Servais, S. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachexia Sarcopenia Muscle 2012, 3, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padrão, A.I.; Oliveira, P.; Vitorino, R.; Colaço, B.; Pires, M.J.; Márquez, M.; Castellanos, E.; Neuparth, M.J.; Teixeira, C.; Costa, C.; et al. Bladder cancer-induced skeletal muscle wasting: Disclosing the role of mitochondria plasticity. Int. J. Biochem. Cell Biol. 2013, 45, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.; Padrão, A.I.; Maciel, E.; Santinha, D.; Oliveira, P.; Vitorino, R.; Moreira-Gonçalves, D.; Colaço, B.; Pires, M.J.; Nunes, C.; et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.M.Y.; Mahendradatta, T.; Taylor, R.J.; Painter, A.B.; Moore, M.M.; Tsoli, M.; Tan, T.C.; Clarke, S.J.; Robertson, G.R.; Polly, P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging 2012, 4, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef]
- Carson, J.A.; Hardee, J.P.; VanderVeen, B.N. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin. Cell Dev. Biol. 2016, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Busquets, S.; Deans, C.; Figueras, M.; Moore-Carrasco, R.; López-Soriano, F.J.; Fearon, K.C.H.; Argilés, J.M. Apoptosis is present in skeletal muscle of cachectic gastro-intestinal cancer patients. Clin. Nutr. 2007, 26, 614–618. [Google Scholar] [CrossRef]
- Prado, C.M.; Lieffers, J.R.; McCargar, L.J.; Reiman, T.; Sawyer, M.B.; Martin, L.; Baracos, V.E. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: A population-based study. Lancet Oncol. 2008, 9, 629–635. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Fainsinger, R.L.; Baracos, V.E.; Md, J.; Loprinzi, C.; Davis, M.; Md, W.; Fearon, K.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Van Dijk, D.P.J.; van de Poll, M.C.G.; Moses, A.G.W.; Preston, T.; Olde Damink, S.W.M.; Rensen, S.S.; Deutz, N.E.P.; Soeters, P.B.; Ross, J.A.; Fearon, K.C.H.; et al. Effects of oral meal feeding on whole body protein breakdown and protein synthesis in cachectic pancreatic cancer patients. J. Cachexia Sarcopenia Muscle 2015, 6, 212–221. [Google Scholar] [CrossRef]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Fanelli, F.R.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.A.; Lin, Y.; Pessin, J.E. Skeletal muscle autophagy: A new metabolic regulator. Trends Endocrinol. Metab. 2013, 24, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Tardif, N.; Klaude, M.; Lundell, L.; Thorell, A.; Rooyackers, O. Autophagic-Lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients 1–3. Am. J. Clin. Nutr. 2013, 98, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Albrecht, D.E.; Lokensgard, B.E.; Spencer, M.J. Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci. 1995, 108 (Pt 6), 2197–2204. [Google Scholar]
- Lee, J.C.; Altschul, R. Electron microscopy of the nuclei of denervated skeletal muscle. Z. Zellforsch. Mikrosk. Anat. 1963, 61, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.O.; Mark Davis, J.; White, J.P.; Carson, J.A. Activity level, apoptosis, and development of cachexia in ApcMin/+ mice. J. Appl. Physiol. 2010, 109, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Belizário, J.E.; Lorite, M.J.; Tisdale, M.J. Cleavage of caspases-1,-3,-6,-8 and -9 substrates by proteases in skeletal muscles from mice undergoing cancer cachexia. Br. J. Cancer 2001, 84, 1135–1140. [Google Scholar] [CrossRef]
- Kinoshita, A.; Onoda, H.; Imai, N.; Iwaku, A.; Oishi, M.; Tanaka, K.; Fushiya, N.; Koike, K.; Nishino, H.; Matsushima, M. The C-Reactive Protein/Albumin Ratio, a Novel Inflammation-Based Prognostic Score, Predicts Outcomes in Patients with Hepatocellular Carcinoma. Ann. Surg. Oncol. 2015, 22, 803–810. [Google Scholar] [CrossRef] [PubMed]
- D’Emilio, A.; Biagiotti, L.; Burattini, S.; Battistelli, M.; Canonico, B.; Evangelisti, C.; Ferri, P.; Papa, S.; Martelli, A.M.; Falcieri, E. Morphological and biochemical patterns in skeletal muscle apoptosis. Histol. Histopathol. 2010, 25, 21–32. [Google Scholar]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Vanderveen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.M.Y.; Poljak, A.; Bentley, N.L.; Turner, N.; Tan, T.C.; Polly, P. Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: Alterations in sarcomeric and mitochondrial protein expression. Oncotarget 2018, 9, 22001–22022. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Age decreases mitochondrial motility and increases mitochondrial size in vascular smooth muscle. J. Physiol. 2016, 594, 4283–4295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leduc-Gaudet, J.-P.; Picard, M.; Pelletier, F.S.-J.; Sgarioto, N.; Auger, M.-J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Lees, H.A.; Manini, T.M.; Buford, T.W.; Aranda, J.M.; Calvani, R.; Capuani, G.; Marsiske, M.; Lott, D.J.; Vandenborne, K.; et al. Skeletal muscle apoptotic signaling predicts thigh muscle volume and gait speed in community-dwelling older persons: An exploratory study. PLoS ONE 2012, 7, e32829. [Google Scholar] [CrossRef] [PubMed]
- Puente-Maestu, L.; Pérez-Parra, J.; Godoy, R.; Moreno, N.; Tejedor, A.; Torres, A.; Lázaro, A.; Ferreira, A.; Agustí, A. Abnormal transition pore kinetics and cytochrome C release in muscle mitochondria of patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 47. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Iqbal, S.; Hood, D.A. The role of mitochondrial fusion and fission in skeletal muscle function and dysfunction. Front. Biosci. 2015, 20, 157–172. [Google Scholar] [Green Version]
- Vitorino, R.; Moreira-Gonçalves, D.; Ferreira, R. Mitochondrial plasticity in cancer-related muscle wasting: Potential approaches for its management. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 2015, 4, e11553. [Google Scholar] [CrossRef] [PubMed]
- Van der Ende, M.; Grefte, S.; Plas, R.; Meijerink, J.; Witkamp, R.F.; Keijer, J.; van Norren, K. Mitochondrial dynamics in cancer-induced cachexia. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 137–150. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+mouse. Skelet. Muscle 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Metzger, G.A.; He, W.A.; Guttridge, D.C. Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J. Cachexia. Sarcopenia Muscle 2014, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Op Den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; De Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.C.; Schols, A.M. Nuclear transcription factor κB activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 2009, 36, 125–131. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Kim, I.; Currin, R.T.; Lemasters, J.J. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2006, 2, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAf-1 cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y. Roles of the Mammalian Mitochondrial Fission and Fusion Mediators Fis1, Drp1, and Opa1 in Apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignotte, B.; Vayssiere, J.L. Mitochondrial control of apoptosis. Adv. Cell Aging Gerontol. 2001, 9, 212–219. [Google Scholar]
- Mignotte, B.; Vayssiere, J.L. Mitochondria and apoptosis. Eur. J. Biochem. 1998, 252, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Harte, M.T.; Kluck, R.M.; Wolf, B.B.; Casiano, C.A.; Newmeyer, D.D.; Wang, H.G.; Reed, J.C.; Nicholson, D.W.; Alnemri, E.S.; et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 1999, 144, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Figueras, M.; Busquets, S.; Carbó, N.; Barreiro, E.; Almendro, V.; Argilés, J.M.; López-Soriano, F.J. Interleukin-15 is able to suppress the increased DNA fragmentation associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2004, 569, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musarò, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Degracia, A.; Busquets, S.; Argilés, J.M.; Bargalló-Gispert, N.; López-Soriano, F.J.; Barreiro, E. Effects of the beta2agonist formoterol on atrophy signaling, autophagy, and muscle phenotype in respiratory and limb muscles of rats with cancer-induced cachexia. Biochimie 2018, 149, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Degracia, A.; Blanco, D.; Vilà-Ubach, M.; de Biurrun, G.; de Solórzano, C.O.; Montuenga, L.M.; Barreiro, E. Phenotypic and metabolic features of mouse diaphragm and gastrocnemius muscles in chronic lung carcinogenesis: Influence of underlying emphysema. J. Transl. Med. 2016, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Mariño, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Lundby, C.; Jacobs, R.A. Adaptations of skeletal muscle mitochondria to exercise training. Exp. Physiol. 2016, 101, 17–22. [Google Scholar] [CrossRef]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Lira, F.S.; Neto, J.C.R.; Seelaender, M. Exercise training as treatment in cancer cachexia. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Métab. 2014, 39, 679–686. [Google Scholar] [CrossRef]
- Spence, R.R.; Heesch, K.C.; Brown, W.J. Exercise and cancer rehabilitation: A systematic review. Cancer Treat. Rev. 2010, 36, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.-H.; Kong, M.-H.; Kim, H.-J. Relationship between low muscle mass and anemia in Korean elderly men: Using the Korea National Health and Nutrition Examination Survey (KNHANES IV–V). J. Clin. Gerontol. Geriatr. 2015, 6, 115–119. [Google Scholar] [CrossRef]
- Cesari, M.; Penninx, B.W.J.H.; Lauretani, F.; Russo, C.R.; Carter, C.; Bandinelli, S.; Atkinson, H.; Onder, G.; Pahor, M.; Ferrucci, L. Hemoglobin levels and skeletal muscle: Results from the InCHIANTI study. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, K.; Stobäus, N.; Reiß, J.; Schulzke, J.; Valentini, L.; Pirlich, M. Effect of sexual dimorphism on muscle strength in cachexia. J. Cachexia Sarcopenia Muscle 2012, 3, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracos, V.E.; Reiman, T.; Mourtzakis, M.; Gioulbasanis, I.; Antoun, S. Body composition in patients with non−small cell lung cancer: A contemporary view of cancer cachexia with the use of computed tomography image analysis. Am. J. Clin. Nutr. 2010, 91, 1133S–1137S. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.C. Systemic inflammation, nutritional status and survival in patients with cancer. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazeby, J.M.; Conroy, T.; Bottomley, A.; Vickery, C.; Arraras, J.; Sezer, O.; Moore, J.; Koller, M.; Turhal, N.S.; Stuart, R.; et al. Clinical and psychometric validation of a questionnaire module, the EORTC QLQ-STO 22, to assess quality of life in patients with gastric cancer. Eur. J. Cancer 2004, 40, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; López-Soriano, F.J.; Toledo, M.; Betancourt, A.; Serpe, R.; Busquets, S. The cachexia score (CASCO): A new tool for staging cachectic cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 87–93. [Google Scholar] [CrossRef]
- Arezzo di Trifiletti, A.; Misino, P.; Giannantoni, P.; Giannantoni, B.; Cascino, A.; Fazi, L.; Rossi Fanelli, F.; Laviano, A. Comparison of the performance of four different tools in diagnosing disease-associated anorexia and their relationship with nutritional, functional and clinical outcome measures in hospitalized patients. Clin. Nutr. 2013, 32, 527–532. [Google Scholar] [CrossRef]
- Rooney, J.P.; Ryde, I.T.; Sanders, L.H.; Howlett, E.H.; Colton, M.D.; Germ, K.E.; Mayer, G.D.; Greenamyre, J.T.; Meyer, J.N. PCR based determination of mitochondrial DNA copy number in multiple species. In Mitochondrial Regulation: Methods and Protocols; Springer: New York, NY, USA, 2014; ISBN 9781493918751. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | WSC | n | CC | n | p Value |
|---|---|---|---|---|---|
| Subjects | 20 | 24 | |||
| Gender (M/F) | 8/12 | 16/8 | |||
| Tumor location (stomach/colorectal) | 6/14 | 8/16 | |||
| Tumor stage | |||||
| I–II | 9 | 4 | |||
| III–IV | 8 | 13 | |||
| Age (years) | 61.00 ± 2.35 | 20 | 61.42 ± 2.68 | 24 | 0.90 |
| Previous weight | 67.50 ± 2.47 | 20 | 73.55 ± 3.40 | 24 | 0.17 |
| Weight (kg) | 65.90 ± 2.79 | 20 | 60.20 ± 2.43 | 24 | 0.13 |
| Height (m) | 1.64 [1.56; 1.70] | 20 | 1.70 [1.64; 1.72] | 24 | 0.14 |
| BMI (kg/m2) | 24.69 ± 0.77 | 20 | 21.55 ± 0.66 | 24 | 0.003 |
| Weight loss (%) | 0.0 [0.0; 0.0] | 20 | 15.12 [8.84; 23.01] | 24 | <0.0001 |
| Hemoglobin (g/dL) | 13.10 [12.20; 14.30] | 14 | 11.30 [9.80; 12.00] | 23 | 0.0001 |
| Serum C reactive protein (CRP) (mg/L) | 5.07 ± 1.29 | 17 | 9.12 ± 0.91 | 22 | 0.01 |
| Serum albumin (mg/dL) | 3.56 ± 0.20 | 17 | 3.25 ± 0.19 | 23 | 0.28 |
| CRP/albumin ratio | 0.18 [0.03; 0.32] | 17 | 0.33 [0.15; 0.42] | 22 | 0.009 |
| Serum glucose (mmol/L) | 5.23 [4.59; 6.20] | 17 | 5.59 [4.75; 6.03] | 22 | 0.67 |
| Serum triacylglycerol (mmol/L) | 1.22 [0.85; 1.75] | 17 | 0.97 [0.81; 1.79] | 22 | 0.60 |
| Serum total cholesterol (mmol/L) | 4.42 [3.54; 5.27] | 16 | 3.41 [2.78; 4.61] | 22 | 0.055 |
| Serum LDL cholesterol (mmol/L) | 2.96 [1.55; 3.41] | 17 | 1.72 [1.29; 2.24] | 21 | 0.11 |
| Serum HDL cholesterol (mmol/L) | 1.04 [0.68; 1.20] | 17 | 0.85 [0.73; 0.98] | 22 | 0.26 |
| Gene | WSC | CC | p Value |
|---|---|---|---|
| Fis1 | 1.06 ± 0.10 | 1.50 ± 0.15 | 0.03 |
| MFN2 | 0.93 [0.71; 1.31] | 1.17 [0.84; 1.86] | 0.20 |
| TFAM | 1.06 [0.78; 1.66] | 1.28 [0.93; 1.71] | 0.38 |
| PGC-1α | 0.97 [0.72; 1.12] | 0.87 [0.57; 1.14] | 0.34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Castro, G.S.; Simoes, E.; Lima, J.D.C.C.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. https://doi.org/10.3390/cancers11091264
de Castro GS, Simoes E, Lima JDCC, Ortiz-Silva M, Festuccia WT, Tokeshi F, Alcântara PS, Otoch JP, Coletti D, Seelaender M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers. 2019; 11(9):1264. https://doi.org/10.3390/cancers11091264
Chicago/Turabian Stylede Castro, Gabriela S., Estefania Simoes, Joanna D.C.C. Lima, Milene Ortiz-Silva, William T. Festuccia, Flávio Tokeshi, Paulo S. Alcântara, José P. Otoch, Dario Coletti, and Marilia Seelaender. 2019. "Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle" Cancers 11, no. 9: 1264. https://doi.org/10.3390/cancers11091264
APA Stylede Castro, G. S., Simoes, E., Lima, J. D. C. C., Ortiz-Silva, M., Festuccia, W. T., Tokeshi, F., Alcântara, P. S., Otoch, J. P., Coletti, D., & Seelaender, M. (2019). Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers, 11(9), 1264. https://doi.org/10.3390/cancers11091264