Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness

 , , and
, , and
Abstract
:1. Introduction
2. Molecular Classification of TNBC
3. Response to Treatment: Clinical Practice and Genomic Alteration of TNBC
4. Current Clinical Trials in Triple-Negative Breast Cancer
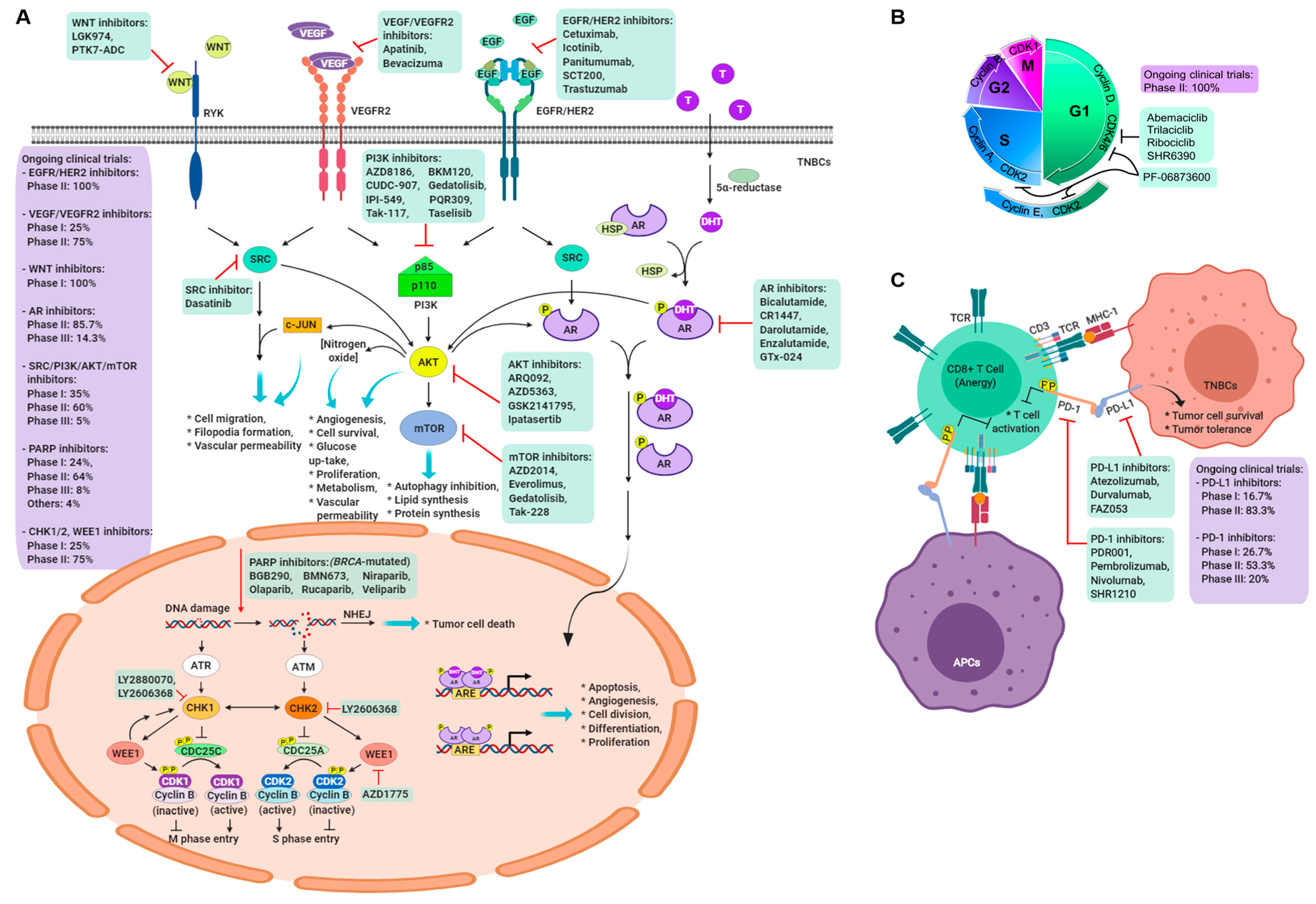
5. Targeting the BRCA1/2 Pathway
5.1. PARP Inhibitors
5.2. Growth Factors and Angiogenesis
5.3. Androgen Receptor Inhibitor
5.4. Immune Checkpoint Molecule Inhibitor
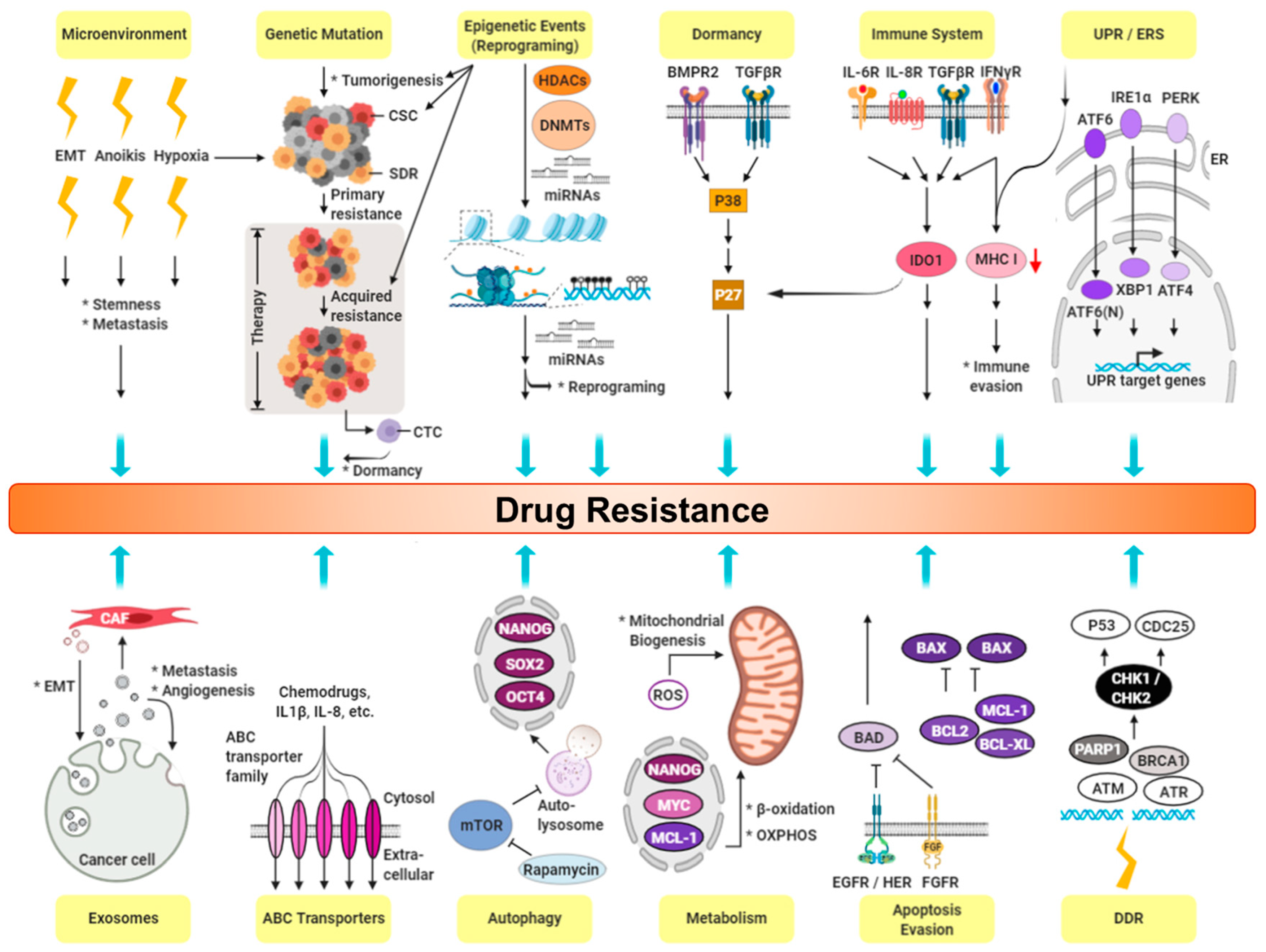
6. CSCs and Drug Resistance in TNBC
6.1. Expression of Cancer Stemness
6.2. Cancer Niche and Therapy Resistance
6.3. Cell Membrane Transporters: ABC Family
6.4. Epithelial-mesenchymal Transition (EMT)
6.5. Hypoxia and ROS
6.6. High Survival Capacity of CSCs
7. Potential Targeting Strategies against CSCs in TNBC
8. Targeting CSCs
8.1. Targeting CSC Specific Marker
8.2. Targeting CSC Signaling
8.3. Targeting CSC Dormancy and Proliferation
8.4. Targeting CSC Metabolism
8.5. Targeting CSC Autophagy
8.6. Reducing CSC Resistance
8.7. Targeting CSC-related Niche
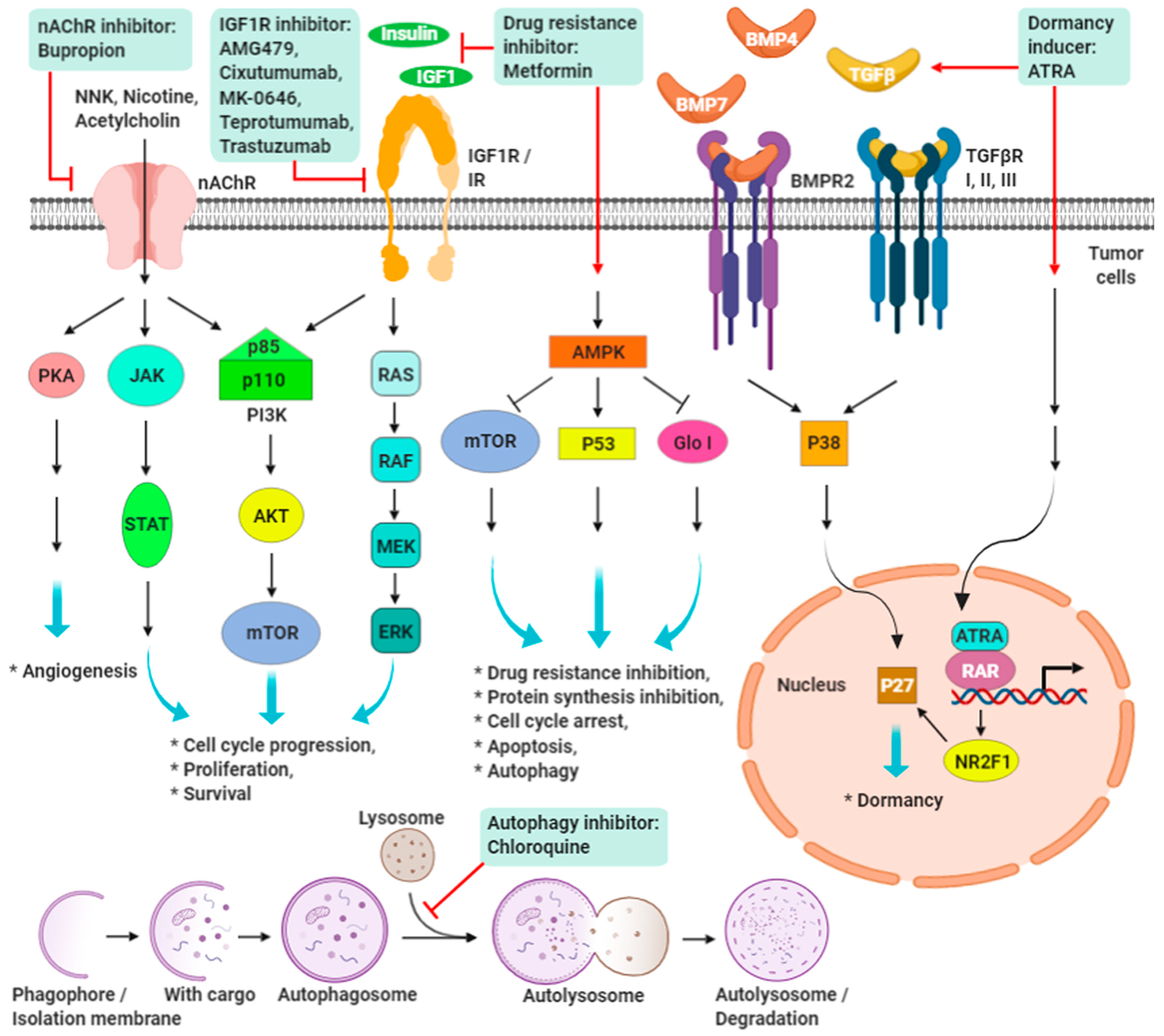
9. Promising Targeting Strategies on IGF-1R and nAChRs in TNBC
9.1. IGF/IGF-1R Signaling
9.2. Nicotine/Nicotinic Acetylcholine Receptors Signaling
10. Precision Treatment of TNBC
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivers, K.F.; Lund, M.J.; Porter, P.L.; Liff, J.M.; Flagg, E.W.; Coates, R.J.; Eley, J.W. The epidemiology of triple-negative breast cancer, including race. Cancer Causes Control 2009, 20, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: literature review. Drug Des. Devel. Ther. 2015, 9, 4303–4318. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.S. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J. Clin. Oncol. 2008, 26, 2223. [Google Scholar] [CrossRef] [PubMed]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome. Biol. 2007, 8, R157. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: a critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Karn, T.; Liedtke, C.; Pusztai, L.; Ruckhaeberle, E.; Hanker, L.; Gaetje, R.; Solbach, C.; Ahr, A.; Metzler, D.; et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011, 13, R97. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Elsawaf, Z.; Sinn, H.P.; Rom, J.; Bermejo, J.L.; Schneeweiss, A.; Aulmann, S. Biological subtypes of triple-negative breast cancer are associated with distinct morphological changes and clinical behaviour. Breast 2013, 22, 986–992. [Google Scholar] [CrossRef]
- Prado-Vazquez, G.; Gamez-Pozo, A.; Trilla-Fuertes, L.; Arevalillo, J.M.; Zapater-Moros, A.; Ferrer-Gomez, M.; Diaz-Almiron, M.; Lopez-Vacas, R.; Navarro, H.; Main, P.; et al. A novel approach to triple-negative breast cancer molecular classification reveals a luminal immune-positive subgroup with good prognoses. Sci. Rep. 2019, 9, 1538. [Google Scholar] [CrossRef]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, B.; Beeler, J.S.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Ashby, W.J.; Lehmann, B.D.; Zijlstra, A.; Pietenpol, J.A.; Moses, H.L. Transforming growth factor beta receptor type III is a tumor promoter in mesenchymal-stem like triple negative breast cancer. Breast Cancer Res. 2014, 16, R69. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Untch, M.; Blohmer, J.U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; Gerber, B.; Eiermann, W.; Hilfrich, J.; Huober, J.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Amir, E.; Clemons, M.; Purdie, C.A.; Miller, N.; Quinlan, P.; Geddie, W.; Coleman, R.E.; Freedman, O.C.; Jordan, L.B.; Thompson, A.M. Tissue confirmation of disease recurrence in breast cancer patients: pooled analysis of multi-centre, multi-disciplinary prospective studies. Cancer Treat. Rev. 2012, 38, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Barni, S. The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res. Treat. 2014, 144, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Graziani, G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol. Res. 2005, 52, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.A.; Lindeman, G.J.; Clemons, M.; Wildiers, H.; Chan, A.; McCarthy, N.J.; Singer, C.F.; Lowe, E.S.; Watkins, C.L.; Carmichael, J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013, 15, R88. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef]
- Palma, J.P.; Rodriguez, L.E.; Bontcheva-Diaz, V.D.; Bouska, J.J.; Bukofzer, G.; Colon-Lopez, M.; Guan, R.; Jarvis, K.; Johnson, E.F.; Klinghofer, V.; et al. The PARP inhibitor, ABT-888 potentiates temozolomide: correlation with drug levels and reduction in PARP activity in vivo. Anticancer Res. 2008, 28, 2625–2635. [Google Scholar]
- Tentori, L.; Muzi, A.; Dorio, A.S.; Scarsella, M.; Leonetti, C.; Shah, G.M.; Xu, W.; Camaioni, E.; Gold, B.; Pellicciari, R.; et al. Pharmacological inhibition of poly(ADP-ribose) polymerase (PARP) activity in PARP-1 silenced tumour cells increases chemosensitivity to temozolomide and to a N3-adenine selective methylating agent. Curr. Cancer Drug Targets 2010, 10, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gomez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Chan, A.; Dirix, L.Y.; Cortes, J.; Pivot, X.; Tomczak, P.; Delozier, T.; Sohn, J.H.; Provencher, L.; Puglisi, F.; et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2010, 28, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Dieras, V.; Cortes, J.; Duenne, A.A.; Yi, J.; O’Shaughnessy, J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: pooled and subgroup analyses of data from 2447 patients. Ann. Oncol. 2013, 24, 2773–2780. [Google Scholar] [CrossRef]
- Bell, R.; Brown, J.; Parmar, M.; Toi, M.; Suter, T.; Steger, G.G.; Pivot, X.; Mackey, J.; Jackisch, C.; Dent, R.; et al. Final efficacy and updated safety results of the randomized phase III BEATRICE trial evaluating adjuvant bevacizumab-containing therapy in triple-negative early breast cancer. Ann. Oncol. 2017, 28, 754–760. [Google Scholar] [CrossRef]
- Brufsky, A.M.; Hurvitz, S.; Perez, E.; Swamy, R.; Valero, V.; O’Neill, V.; Rugo, H.S. RIBBON-2: a randomized, double-blind, placebo-controlled, phase III trial evaluating the efficacy and safety of bevacizumab in combination with chemotherapy for second-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2011, 29, 4286–4293. [Google Scholar]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef]
- Collins, L.C.; Cole, K.S.; Marotti, J.D.; Hu, R.; Schnitt, S.J.; Tamimi, R.M. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod. Pathol. 2011, 24, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; ME, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2019, (in press). [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatina, J. The dynamics of cancer stem cells. Neoplasma 2012, 59, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arques, O.; Martinez-Quintanilla, J.; Cuesta-Borras, E.; Ramirez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Invest. 2018, 128, 3887–3905. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yoshioka, Y.; Minoura, K.; Takahashi, R.U.; Takeshita, F.; Taya, T.; Horii, R.; Fukuoka, Y.; Kato, T.; Kosaka, N.; et al. An integrative genomic analysis revealed the relevance of microRNA and gene expression for drug-resistance in human breast cancer cells. Mol. Cancer 2011, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Efferth, T. Tumor Heterogeneity, Single-Cell Sequencing, and Drug Resistance. Pharmaceuticals 2016, 9, 33. [Google Scholar] [CrossRef]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.; Volm, M. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Naik, P.P.; Das, D.N.; Panda, P.K.; Mukhopadhyay, S.; Sinha, N.; Praharaj, P.P.; Agarwal, R.; Bhutia, S.K. Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral. Oncol. 2016, 62, 122–135. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.T.; ME, L.L. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 2016, 49, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G., 2nd; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitao, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, J.; Jiang, Y.; Yu, C.; Ma, Z. Predictive value of CD44 and CD24 for prognosis and chemotherapy response in invasive breast ductal carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 11287–11295. [Google Scholar]
- Lin, Y.; Zhong, Y.; Guan, H.; Zhang, X.; Sun, Q. CD44+/CD24- phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. J. Exp. Clin. Cancer Res. 2012, 31, 59. [Google Scholar] [CrossRef]
- Li, H.; Ma, F.; Wang, H.; Lin, C.; Fan, Y.; Zhang, X.; Qian, H.; Xu, B. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int. J. Biol. Markers 2013, 28, e357–364. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Li, Y.; Ding, X.; Wang, H.; Fan, Y.; Lin, C.; Qian, H.; Xu, B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine 2017, 96, e6561. [Google Scholar] [CrossRef]
- Ohi, Y.; Umekita, Y.; Yoshioka, T.; Souda, M.; Rai, Y.; Sagara, Y.; Sagara, Y.; Sagara, Y.; Tanimoto, A. Aldehyde dehydrogenase 1 expression predicts poor prognosis in triple-negative breast cancer. Histopathology 2011, 59, 776–780. [Google Scholar] [CrossRef]
- Moreira, M.P.; da Conceicao Braga, L.; Cassali, G.D.; Silva, L.M. STAT3 as a promising chemoresistance biomarker associated with the CD44(+/high)/CD24(-/low)/ALDH(+) BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp. Cell Res. 2018, 363, 283–290. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Goldberg, A.; Lin, Z.; Ko, Y.H.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol. Ther. 2011, 12, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, M.; Kruger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef]
- Rafii, A.; Mirshahi, P.; Poupot, M.; Faussat, A.M.; Simon, A.; Ducros, E.; Mery, E.; Couderc, B.; Lis, R.; Capdet, J.; et al. Oncologic trogocytosis of an original stromal cells induces chemoresistance of ovarian tumours. PLoS ONE 2008, 3, e3894. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.S.; Rustgi, A.K. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br. J. Cancer 2013, 108, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, T. Lipid Raft-Mediated Regulation of Hyaluronan-CD44 Interactions in Inflammation and Cancer. Front. Immunol. 2015, 6, 420. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport--an update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Chuthapisith, S.; Eremin, J.; El-Sheemey, M.; Eremin, O. Breast cancer chemoresistance: emerging importance of cancer stem cells. Surg. Oncol. 2010, 19, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; Foekens, J.A.; Look, M.P.; Meijer-van Gelder, M.E.; Klijn, J.G.; Wiemer, E.A.; Stoter, G.; Nooter, K. RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: correlation with chemotherapeutic response. Clin. Cancer Res. 2003, 9, 827–836. [Google Scholar] [PubMed]
- Hawley, T.S.; Riz, I.; Yang, W.; Wakabayashi, Y.; Depalma, L.; Chang, Y.T.; Peng, W.; Zhu, J.; Hawley, R.G. Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am. J. Hematol. 2013, 88, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Bradford, G.B.; Williams, B.; Rossi, R.; Bertoncello, I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp. Hematol. 1997, 25, 445–453. [Google Scholar]
- Zhu, L.F.; Hu, Y.; Yang, C.C.; Xu, X.H.; Ning, T.Y.; Wang, Z.L.; Ye, J.H.; Liu, L.K. Snail overexpression induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. Lab. Invest. 2012, 92, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Au, H.K.; Hsu, J.L.; Wang, H.F.; Lee, C.J.; Peng, S.W.; Lai, S.C.; Wu, Y.C.; Ho, H.N.; Huang, Y.H. IGF-1R Promotes Symmetric Self-Renewal and Migration of Alkaline Phosphatase(+) Germ Stem Cells through HIF-2alpha-OCT4/CXCR4 Loop under Hypoxia. Stem Cell Reports 2018, 10, 524–537. [Google Scholar] [CrossRef]
- Das, B.; Tsuchida, R.; Malkin, D.; Koren, G.; Baruchel, S.; Yeger, H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 2008, 26, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Schwab, L.P.; Peacock, D.L.; Majumdar, D.; Ingels, J.F.; Jensen, L.C.; Smith, K.D.; Cushing, R.C.; Seagroves, T.N. Hypoxia-inducible factor 1alpha promotes primary tumor growth and tumor-initiating cell activity in breast cancer. Breast Cancer Res. 2012, 14, R6. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, D.H.; Landon, C.D.; Nien, C.Y.; Hanna, G.; Rich, J.N.; Provenzale, J.M.; et al. Tumor cells upregulate normoxic HIF-1alpha in response to doxorubicin. Cancer Res. 2013, 73, 6230–6242. [Google Scholar] [CrossRef]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Salnikov, A.V.; Bauer, N.; Aleksandrowicz, E.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Schemmer, P.; Werner, J.; et al. Triptolide reverses hypoxia-induced epithelial-mesenchymal transition and stem-like features in pancreatic cancer by NF-kappaB downregulation. Int. J. Cancer 2014, 134, 2489–2503. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Almog, N. Molecular mechanisms underlying tumor dormancy. Cancer Lett 2010, 294, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Li, X.S.; Xu, Q.; Fu, X.Y.; Luo, W.S. ALDH1A1 overexpression is associated with the progression and prognosis in gastric cancer. BMC Cancer 2014, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Balicki, D. Moving forward in human mammary stem cell biology and breast cancer prognostication using ALDH1. Cell Stem Cell 2007, 1, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Huo, W.; Du, M.; Pan, X.; Zhu, X.; Li, Z. Prognostic value of ALDH1 expression in lung cancer: a meta-analysis. Int J. Clin. Exp. Med. 2015, 8, 2045–2051. [Google Scholar] [PubMed]
- Ikeda, J.; Mamat, S.; Tian, T.; Wang, Y.; Luo, W.; Rahadiani, N.; Aozasa, K.; Morii, E. Reactive oxygen species and aldehyde dehydrogenase activity in Hodgkin lymphoma cells. Lab. Invest. 2012, 92, 606–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Smith, C.A. ALDHs in normal and malignant hematopoietic cells: Potential new avenues for treatment of AML and other blood cancers. Chem. Biol. Interact. 2017, 276, 46–51. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Kida, K.; Ishikawa, T.; Yamada, A.; Shimada, K.; Narui, K.; Sugae, S.; Shimizu, D.; Tanabe, M.; Sasaki, T.; Ichikawa, Y.; et al. Effect of ALDH1 on prognosis and chemoresistance by breast cancer subtype. Breast Cancer Res. Treat. 2016, 156, 261–269. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010, 12, R31. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Zhang, M.; Rajapakshe, K.; Coarfa, C.; Edwards, D.; Huang, S.; Rosen, J.M. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Reports 2015, 5, 378–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Atkinson, R.L.; Rosen, J.M. Selective targeting of radiation-resistant tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3522–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreb, J.S.; Mohuczy, D.; Ostmark, B.; Zucali, J.R. RNAi-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother. Pharmacol. 2007, 59, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Aires, A.; Ocampo, S.M.; Simoes, B.M.; Josefa Rodriguez, M.; Cadenas, J.F.; Couleaud, P.; Spence, K.; Latorre, A.; Miranda, R.; Somoza, A.; et al. Multifunctionalized iron oxide nanoparticles for selective drug delivery to CD44-positive cancer cells. Nanotechnology 2016, 27, 065103. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, E.M.F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Mamaeva, V.; Niemi, R.; Beck, M.; Ozliseli, E.; Desai, D.; Landor, S.; Gronroos, T.; Kronqvist, P.; Pettersen, I.K.; McCormack, E.; et al. Inhibiting Notch Activity in Breast Cancer Stem Cells by Glucose Functionalized Nanoparticles Carrying gamma-secretase Inhibitors. Mol. Ther. 2016, 24, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roche, H.; Sparano, J.; Strauss, L.C.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin. Cancer Res. 2011, 17, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Solzak, J.P.; Atale, R.V.; Hancock, B.A.; Sinn, A.L.; Pollok, K.E.; Jones, D.R.; Radovich, M. Dual PI3K and Wnt pathway inhibition is a synergistic combination against triple negative breast cancer. NPJ Breast Cancer 2017, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Abderrahman, B.; Jordan, V.C. Rethinking Extended Adjuvant Antiestrogen Therapy to Increase Survivorship in Breast Cancer. JAMA Oncol. 2018, 4, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sanchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Altman, J.K. How I treat acute promyelocytic leukemia. Blood 2009, 114, 5126–5135. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, Z.; Xu, X.; Chen, C.; Wei, W.; Fan, M.; Chen, X.; Li, J.J.; Wang, Y.; Huang, J. All-trans retinoic acids induce differentiation and sensitize a radioresistant breast cancer cells to chemotherapy. BMC Complement. Altern. Med. 2016, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, N.; Lin, W.C.; Wehde, B.L.; Triplett, A.A.; Wagner, K.U. Autocrine IGF1 Signaling Mediates Pancreatic Tumor Cell Dormancy in the Absence of Oncogenic Drivers. Cell Rep. 2017, 18, 2243–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; Hulver, M.W.; Schmelz, E.M. Ovarian tumor-initiating cells display a flexible metabolism. Exp. Cell Res. 2014, 328, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Suva, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clement-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyari, Y.; Dratver, M.B.; Pajonk, F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014, 146, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annibaldi, A.; Widmann, C. Glucose metabolism in cancer cells. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Dmitriev, A.A.; Snezhkina, A.V.; Kudryavtseva, A.V. Deregulation of glycolysis in cancer: glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target. Expert Opin. Ther. Targets 2013, 17, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Gentles, A.; Nair, R.V.; Huang, M.; Lin, Y.; Lee, C.Y.; Cai, S.; Scheeren, F.A.; Kuo, A.H.; Diehn, M. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells 2014, 32, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef]
- Serafino, A.; Zonfrillo, M.; Andreola, F.; Psaila, R.; Mercuri, L.; Moroni, N.; Renier, D.; Campisi, M.; Secchieri, C.; Pierimarchi, P. CD44-targeting for antitumor drug delivery: a new SN-38-hyaluronan bioconjugate for locoregional treatment of peritoneal carcinomatosis. Curr. Cancer Drug Targets 2011, 11, 572–585. [Google Scholar] [CrossRef]
- Tamada, M.; Nagano, O.; Tateyama, S.; Ohmura, M.; Yae, T.; Ishimoto, T.; Sugihara, E.; Onishi, N.; Yamamoto, T.; Yanagawa, H.; et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 2012, 72, 1438–1448. [Google Scholar] [CrossRef]
- Dupuy, F.; Tabaries, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Chaterjee, M.; van Golen, K.L. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011, 2011, 362938. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, Q.; Sun, H.; Yin, L.; Wu, J.; Xu, J.; He, T.; Yang, C.; Liang, C. Defective autophagy leads to the suppression of stem-like features of CD271(+) osteosarcoma cells. J. Biomed. Sci. 2016, 23, 82. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Gong, C.; Song, E.; Codogno, P.; Mehrpour, M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy 2012, 8, 1853–1855. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfbeta/Smad Signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Ghezzi, P.; Rumio, C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol. Cancer 2017, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Shen, S.; Zhang, Y.J.; Xu, C.F.; Cao, Z.T.; Wen, L.P.; Wang, J. Nanoparticle-facilitated autophagy inhibition promotes the efficacy of chemotherapeutics against breast cancer stem cells. Biomaterials 2016, 103, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, J.; Liu, Z.; Pan, L.; Xu, G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol. Lett. 2018, 15, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Del Barco, S.; Vazquez-Martin, A.; Cufi, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Takeshita, F.; Yamamoto, Y.; Minoura, K.; Ono, M.; Kodaira, M.; Tamura, K.; Mori, M.; Ochiya, T. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat. Commun. 2015, 6, 7318. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Liu, W.; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017, 3, 17010. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Agbor-Tarh, D.; Bradbury, I.; Di Cosimo, S.; Azim, H.A., Jr.; Fumagalli, D.; Sarp, S.; Wolff, A.C.; Andersson, M.; Kroep, J.; et al. Impact of Diabetes, Insulin, and Metformin Use on the Outcome of Patients With Human Epidermal Growth Factor Receptor 2-Positive Primary Breast Cancer: Analysis From the ALTTO Phase III Randomized Trial. J. Clin. Oncol. 2017, 35, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Son, K.; Fujioka, S.; Iida, T.; Furukawa, K.; Fujita, T.; Yamada, H.; Chiao, P.J.; Yanaga, K. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer Res. 2009, 29, 3995–4003. [Google Scholar] [PubMed]
- Duivenvoorden, W.C.; Popovic, S.V.; Lhotak, S.; Seidlitz, E.; Hirte, H.W.; Tozer, R.G.; Singh, G. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer Res. 2002, 62, 1588–1591. [Google Scholar] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting hypoxic cancer stem cells (CSCs) with Doxycycline: Implications for optimizing anti-angiogenic therapy. Oncotarget 2017, 8, 56126–56142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef]
- Yang, W.C.; Chen, H.W.; Au, H.K.; Chang, C.W.; Huang, C.T.; Yen, Y.H.; Tzeng, C.R. Serum and endometrial markers. Best Pract Res. Clin. Obstet. Gynaecol. 2004, 18, 305–318. [Google Scholar] [CrossRef]
- Au, H.K.; Chang, J.H.; Wu, Y.C.; Kuo, Y.C.; Chen, Y.H.; Lee, W.C.; Chang, T.S.; Lan, P.C.; Kuo, H.C.; Lee, K.L.; et al. TGF-betaI Regulates Cell Migration through Pluripotent Transcription Factor OCT4 in Endometriosis. PLoS ONE 2015, 10, e0145256. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Chen, C.L.; Wu, Y.C.; Liu, J.J.; Kuo, Y.C.; Lee, K.F.; Lin, S.Y.; Lin, S.E.; Tung, S.Y.; Kuo, L.M.; et al. Inflammation Promotes Expression of Stemness-Related Properties in HBV-Related Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0149897. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Wu, Y.C.; Chi, C.C.; Su, W.C.; Chang, P.J.; Lee, K.F.; Tung, T.H.; Wang, J.; Liu, J.J.; Tung, S.Y.; et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin. Cancer Res. 2015, 21, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Lin, M.H.; Wang, P.C.; Wu, Y.C.; Chiang, H.L.; Wang, Y.L.; Chang, J.H.; Huang, Y.K.; Gu, S.Y.; Ho, H.N.; et al. Hypoxia inducible factor 2alpha/insulin-like growth factor receptor signal loop supports the proliferation and Oct-4 maintenance of mouse germline stem cells. Mol. Hum. Reprod. 2014, 20, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Yu, J.; Cheng, A.S.; Wong, G.L.; Chan, H.Y.; Chu, E.S.; Ng, E.K.; Chan, F.K.; Sung, J.J.; Chan, H.L. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int. J. Cancer 2009, 124, 2766–2770. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.N.; Pan, J.J.; Huang, Y.W.; Tsai, H.J.; Chang, W.C. Association between Nonsteroidal Anti-Inflammatory Drugs and Colorectal Cancer: A Population-Based Case-Control Study. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.M.; Kwon, J.H.; Kim, J.S.; Oh, S.H.; Jin Lee, K.; Park, J.J.; Pil Hong, S.; Cheon, J.H.; Kim, T.I.; Kim, W.H. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int. J. Cancer 2014, 134, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.; Penarando, J.; Canas, A.; Lopez-Sanchez, L.M.; Conde, F.; Hernandez, V.; Peralbo, E.; Lopez-Pedrera, C.; de la Haba-Rodriguez, J.; Aranda, E.; et al. Simultaneous inhibition of EGFR/VEGFR and cyclooxygenase-2 targets stemness-related pathways in colorectal cancer cells. PLoS ONE 2015, 10, e0131363. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.R.; Cheon, H.; Junk, D.J.; Vinayak, S.; Varadan, V.; Telli, M.L.; Ford, J.M.; Stark, G.R.; Jackson, M.W. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13792–13797. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sancho, P.; Canamero, M.; Martinelli, P.; Madriles, F.; Michl, P.; Gress, T.; de Pascual, R.; Gandia, L.; Guerra, C.; et al. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 2014, 147, 1119–1133 e1114. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.M.; Bora-Singhal, N.; Kumar, D.M.; Chellappan, S.P. Regulation of Sox2 and stemness by nicotine and electronic-cigarettes in non-small cell lung cancer. Mol. Cancer 2018, 17, 149. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G. Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer. J. Clin. Oncol. 2010, 28, 4985–4995. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Stewart, M.H.; Menendez, P.; George, D.; Vijayaragavan, K.; Werbowetski-Ogilvie, T.; Ramos-Mejia, V.; Rouleau, A.; Yang, J.; Bosse, M.; et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 2007, 448, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic-Arsic, M.; Kalideris, E.; Siveke, J.T. The role of insulin and IGF system in pancreatic cancer. J. Mol. Endocrinol. 2013, 50, R67–R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.J.; Ying, X.J.; Chen, H.L.; Ye, P.J.; Chen, Z.L.; Li, G.; Jiang, H.F.; Liu, J.; Zhou, S.Z. Insulin-like growth factor-1 induces lymphangiogenesis and facilitates lymphatic metastasis in colorectal cancer. World J. Gastroenterol. 2013, 19, 7788–7794. [Google Scholar] [CrossRef] [PubMed]
- Kubasiak, J.C.; Seder, C.W.; Pithadia, R.; Basu, S.; Fhied, C.; Phillips, W.W.; Daly, S.; Shersher, D.D.; Yoder, M.A.; Chmielewski, G.; et al. Value of circulating insulin-like growth factor-associated proteins for the detection of stage I non-small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2015, 149, 727–734 e721-723, discussion 734. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Cheng, W.F.; Wu, Y.P.; Cheng, Y.M.; Hsu, K.F.; Chou, C.Y. Circulating IGF system and treatment outcome in epithelial ovarian cancer. Endocr. Relat. Cancer 2014, 21, 217–229. [Google Scholar] [CrossRef]
- Cao, Y.; Lindstrom, S.; Schumacher, F.; Stevens, V.L.; Albanes, D.; Berndt, S.; Boeing, H.; Bueno-de-Mesquita, H.B.; Canzian, F.; Chamosa, S.; et al. Insulin-like growth factor pathway genetic polymorphisms, circulating IGF1 and IGFBP3, and prostate cancer survival. J. Natl. Cancer Inst. 2014, 106, dju085. [Google Scholar] [CrossRef]
- Bruchim, I.; Sarfstein, R.; Werner, H. The IGF Hormonal Network in Endometrial Cancer: Functions, Regulation, and Targeting Approaches. Front. Endocrinol. (Lausanne) 2014, 5, 76. [Google Scholar] [CrossRef] [Green Version]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Tsao, S.W.; Chan, K.W.; Ludwig, D.L.; Novosyadlyy, R.; Li, Y.Y.; He, Q.Y.; Cheung, A.L. Id1-induced IGF-II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance--implications for IGF-II and IGF-IR-targeted therapy. Clin. Cancer Res. 2014, 20, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Gaikwad, S.M.; Jinager, A.; Chaudhury, S.; Maheshwari, A.; Ray, P. IGF-1R inhibition potentiates cytotoxic effects of chemotherapeutic agents in early stages of chemoresistant ovarian cancer cells. Cancer Lett. 2014, 354, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Cerna, M.; Narsanska, A.; Treska, V.; Kucera, R.; Topolcan, O. IGF1 and tumor markers in different breast cancer stages. Rozhl. Chir. 2011, 90, 688–694. [Google Scholar] [PubMed]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.; Hilsenbeck, S.G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Chang, J.C.; et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.W.; Lin, R.J.; Yu, J.; Chang, W.Y.; Fu, C.H.; Lai, A.; Yu, J.C.; Yu, A.L. The expression and significance of insulin-like growth factor-1 receptor and its pathway on breast cancer stem/progenitors. Breast Cancer Res. 2013, 15, R39. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Dhadve, A.; Sakpal, A.; De, A.; Ray, P. An active IGF-1R-AKT signaling imparts functional heterogeneity in ovarian CSC population. Sci. Rep. 2016, 6, 36612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Chang, Y.C.; Chen, C.S.; Tu, S.H.; Wang, Y.J.; Chen, L.C.; Chang, Y.J.; Wei, P.L.; Chang, H.W.; Chang, C.H.; et al. Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces alpha9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res. Treat. 2011, 129, 331–345. [Google Scholar] [CrossRef]
- Aali, N.; Motalleb, G. The effect of nicotine on the expressions of the alpha7 nicotinic receptor gene and Bax and Bcl-2 proteins in the mammary gland epithelial-7 breast cancer cell line and its relationship to drug resistance. Cell Mol. Biol. Lett. 2015, 20, 948–964. [Google Scholar] [CrossRef]
- Davis, R.; Rizwani, W.; Banerjee, S.; Kovacs, M.; Haura, E.; Coppola, D.; Chellappan, S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 2009, 4, e7524. [Google Scholar] [CrossRef]
- Heeschen, C.; Jang, J.J.; Weis, M.; Pathak, A.; Kaji, S.; Hu, R.S.; Tsao, P.S.; Johnson, F.L.; Cooke, J.P. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 2001, 7, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Huang, C.S.; Chen, C.S.; Tu, S.H.; Wang, Y.J.; Chang, Y.J.; Tam, K.W.; Wei, P.L.; Cheng, T.C.; Chu, J.S.; et al. Overexpression and activation of the alpha9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J. Natl. Cancer Inst. 2010, 102, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Ho, Y.S.; Guo, H.R.; Wang, Y.J. Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol. Sci. 2008, 104, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Kim, H.S.; Luo, L.Y.; Huang, Y.; Guo, J.; Chen, C.Y. Sensitization of epithelial growth factor receptors by nicotine exposure to promote breast cancer cell growth. Breast Cancer Res. 2011, 13, R113. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Chen, C.H.; Wang, Y.J.; Pestell, R.G.; Albanese, C.; Chen, R.J.; Chang, M.C.; Jeng, J.H.; Lin, S.Y.; Liang, Y.C.; et al. Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFkappaB activation and cyclin D1 up-regulation. Toxicol. Appl. Pharmacol. 2005, 205, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Chellappan, S. alpha7 nicotinic acetylcholine receptor subunit in angiogenesis and epithelial to mesenchymal transition. Curr. Drug Targets 2012, 13, 671–679. [Google Scholar] [PubMed]
- Lien, Y.C.; Wang, W.; Kuo, L.J.; Liu, J.J.; Wei, P.L.; Ho, Y.S.; Ting, W.C.; Wu, C.H.; Chang, Y.J. Nicotine promotes cell migration through alpha7 nicotinic acetylcholine receptor in gastric cancer cells. Ann. Surg. Oncol. 2011, 18, 2671–2679. [Google Scholar] [CrossRef] [PubMed]
- Momi, N.; Ponnusamy, M.P.; Kaur, S.; Rachagani, S.; Kunigal, S.S.; Chellappan, S.; Ouellette, M.M.; Batra, S.K. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through alpha7nAChR-mediated MUC4 upregulation. Oncogene 2013, 32, 1384–1395. [Google Scholar] [CrossRef]
- Wang, W.; Chin-Sheng, H.; Kuo, L.J.; Wei, P.L.; Lien, Y.C.; Lin, F.Y.; Liu, H.H.; Ho, Y.S.; Wu, C.H.; Chang, Y.J. NNK enhances cell migration through alpha7-nicotinic acetylcholine receptor accompanied by increased of fibronectin expression in gastric cancer. Ann. Surg. Oncol. 2012, 19, S580–S588. [Google Scholar] [CrossRef]
- Wei, P.L.; Chang, Y.J.; Ho, Y.S.; Lee, C.H.; Yang, Y.Y.; An, J.; Lin, S.Y. Tobacco-specific carcinogen enhances colon cancer cell migration through alpha7-nicotinic acetylcholine receptor. Ann. Surg. 2009, 249, 978–985. [Google Scholar] [CrossRef]
- Chen, R.J.; Ho, Y.S.; Guo, H.R.; Wang, Y.J. Long-term nicotine exposure-induced chemoresistance is mediated by activation of Stat3 and downregulation of ERK1/2 via nAChR and beta-adrenoceptors in human bladder cancer cells. Toxicol. Sci. 2010, 115, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Trevino, J.G.; Pillai, S.; Kunigal, S.; Singh, S.; Fulp, W.J.; Centeno, B.A.; Chellappan, S.P. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia 2012, 14, 1102–1114. [Google Scholar] [CrossRef]
- Guha, P.; Bandyopadhyaya, G.; Polumuri, S.K.; Chumsri, S.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Nicotine promotes apoptosis resistance of breast cancer cells and enrichment of side population cells with cancer stem cell-like properties via a signaling cascade involving galectin-3, alpha9 nicotinic acetylcholine receptor and STAT3. Breast Cancer Res. Treat. 2014, 145, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, M.H.; Banerjee, J.; Al-Wadei, H.A.; Schuller, H.M. Nicotine induces self-renewal of pancreatic cancer stem cells via neurotransmitter-driven activation of sonic hedgehog signalling. Eur. J. Cancer 2016, 52, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Chang, Y.C. Enhancement of cancer stem-like and epithelial-mesenchymal transdifferentiation property in oral epithelial cells with long-term nicotine exposure: reversal by targeting SNAIL. Toxicol. Appl. Pharmacol. 2013, 266, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Sekino, Y.; Kanda, Y. Nicotine increases cancer stem cell population in MCF-7 cells. Biochem. Biophys. Res. Commun. 2010, 403, 138–143. [Google Scholar] [CrossRef]
- Dinicola, S.; Morini, V.; Coluccia, P.; Proietti, S.; D’Anselmi, F.; Pasqualato, A.; Masiello, M.G.; Palombo, A.; De Toma, G.; Bizzarri, M.; et al. Nicotine increases survival in human colon cancer cells treated with chemotherapeutic drugs. Toxicol. In Vitro 2013, 27, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Cucina, A.; Dinicola, S.; Coluccia, P.; Proietti, S.; D’Anselmi, F.; Pasqualato, A.; Bizzarri, M. Nicotine stimulates proliferation and inhibits apoptosis in colon cancer cell lines through activation of survival pathways. J. Surg. Res. 2012, 178, 233–241. [Google Scholar] [CrossRef]
- Wong, H.P.; Yu, L.; Lam, E.K.; Tai, E.K.; Wu, W.K.; Cho, C.H. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol. Appl. Pharmacol. 2007, 221, 261–267. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Lee, C.H.; Tu, S.H.; Wu, C.H.; Hung, C.S.; Hsieh, M.C.; Chuang, C.W.; Ho, Y.S.; Chiou, H.Y. CHRNA9 polymorphisms and smoking exposure synergize to increase the risk of breast cancer in Taiwan. Carcinogenesis 2014, 35, 2520–2525. [Google Scholar] [CrossRef] [Green Version]
- Valor, L.M.; Castillo, M.; Ortiz, J.A.; Criado, M. Transcriptional regulation by activation and repression elements located at the 5’-noncoding region of the human alpha9 nicotinic receptor subunit gene. J. Biol. Chem. 2003, 278, 37249–37255. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lee, C.H.; Chuang, Y.H.; Lee, J.Y.; Chiu, Y.Y.; Wu Lee, Y.H.; Jong, Y.J.; Hwang, J.K.; Huang, S.H.; Chen, L.C.; et al. Membrane protein-regulated networks across human cancers. Nat. Commun. 2019, 10, 3131. [Google Scholar] [CrossRef] [PubMed]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Brough, R.; Frankum, J.R.; Sims, D.; Mackay, A.; Mendes-Pereira, A.M.; Bajrami, I.; Costa-Cabral, S.; Rafiq, R.; Ahmad, A.S.; Cerone, M.A.; et al. Functional viability profiles of breast cancer. Cancer Discov. 2011, 1, 260–273. [Google Scholar] [CrossRef]
- Daemen, A.; Griffith, O.L.; Heiser, L.M.; Wang, N.J.; Enache, O.M.; Sanborn, Z.; Pepin, F.; Durinck, S.; Korkola, J.E.; Griffith, M.; et al. Modeling precision treatment of breast cancer. Genome. Biol. 2013, 14, R110. [Google Scholar] [CrossRef]
- Gautam, P.; Karhinen, L.; Szwajda, A.; Jha, S.K.; Yadav, B.; Aittokallio, T.; Wennerberg, K. Identification of selective cytotoxic and synthetic lethal drug responses in triple negative breast cancer cells. Mol. Cancer 2016, 15, 34. [Google Scholar] [CrossRef]
- Gautam, P.; Jaiswal, A.; Aittokallio, T.; Al-Ali, H.; Wennerberg, K. Phenotypic Screening Combined with Machine Learning for Efficient Identification of Breast Cancer-Selective Therapeutic Targets. Cell Chem. Biol. 2019, 26, 970–979. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Major Drug | Combinational Drug | Phase | NCT Identifiers | Status # |
|---|---|---|---|---|---|
| BRCA1/2 | |||||
| PARP | BGB290 | ± Temozolomide | I/II | NCT03150810 | R |
| BMN-673 | - | II | NCT02401347 | R | |
| ± Carboplatin + Paclitaxel | I | NCT02358200 | Ac/NR | ||
| ± CB-839 (GLS1i) | I/II | NCT03875313 | R | ||
| ± ZEN003694 (BETi) | II | NCT03901469 | R | ||
| Niraparib | ± Pembrolizumab | I/II | NCT02657889 | Ac/NR | |
| Olaparib | - | II | NCT00679783 | Ac/NR | |
| ± AZD2014 (mTORi) ± AZD5363 (AKTi) | I/II | NCT02208375 | Ac/NR | ||
| ± AZD6738 ± AZD1775 | II | NCT03330847 | R | ||
| ± BKM120 (PI3Ki) ± BYL719 (PI3Ki) | I | NCT01623349 | Ac/NR | ||
| ± Carboplatin ± Paclitaxel | I | NCT00516724 | Ac/NR | ||
| ± Carboplatin ± Paclitaxel | II/III | NCT03150576 | R | ||
| ± Cediranib (VGEFi) | I/II | NCT01116648 | Ac/NR | ||
| ± Cediranib ± Durvalumab | I/II | NCT02484404 | R | ||
| ± Durvalumab (anti PD-1) | I | NCT03544125 | R | ||
| ± Durvalumab (anti PD-1) | II | NCT03801369 | R | ||
| ± Durvalumab (anti PD-1) | II | NCT03167619 | R | ||
| ± Onalespib (HSP90i) | I | NCT02898207 | R | ||
| Rucaparib | ± Cisplatin | II | NCT01074970 | Ac/NR | |
| Veliparib | ± Carboplatin ± Cyclophosphamide ± Doxorubicin ± Paclitaxel | II | NCT01818063 | Ac/NR | |
| ± Carboplatin ± Cyclophosphamide ± Doxorubicin ± Paclitaxel | III | NCT02032277 | Ac/NR | ||
| ± Cisplatin | II | NCT02595905 | R | ||
| ± Irinotecan Hydrochloride | I | NCT00576654 | Ac/NR | ||
| ± Lapatinib (EGFR/HER2i) | Pilot study | NCT02158507 | Ac/NR | ||
| CHK1 and WEE1 | |||||
| CHK1 | LY2880070 | ± Gemcitabine | I /II | NCT02632448 | R |
| CHK1/2 | LY2606368 | - | II | NCT02203513 | R |
| WEE1 | AZD1775 | - | I | NCT02482311 | Ac/NR |
| ± Cisplatin | II | NCT03012477 | Ac/NR | ||
| Cyclin-dependent kinases | |||||
| CDKs | Abemaciclib | - | II | NCT03130439 | R |
| PF-06873600 | ± Endocrine Therapy | II | NCT03519178 | R | |
| Ribociclib | ± Bicalutamide (ARi) | I/II | NCT03090165 | Ac/NR | |
| SHR6390 | ± SHR3680 (ARi) | I/II | NCT03805399 | R | |
| Trilaciclib | ± Gemcitabine + Carboplatin | II | NCT02978716 | Ac/NR | |
| Androgen receptor | |||||
| Androgen | Bicalutamide | - | III | NCT03055312 | R |
| CR1447(ARM) | - | II | NCT02067741 | Ac/NR | |
| Darolutamide (NSAA) | ± Capecitabine | II | NCT03383679 | R | |
| Enzalutamide | - | II | NCT02750358 | Ac/NR | |
| - | II | NCT01889238 | Ac/NR | ||
| ± Paclitaxel | II | NCT02689427 | R | ||
| GTx-024 (SARM) | ± Pambrolizumab (anti PD-1) | II | NCT02971761 | R | |
| Growth factors and angiogenesis | |||||
| EGFR | Cetuximab | ± Ixabepilone | II | NCT01097642 | Ac/NR |
| Icotinib | - | II | NCT02362230 | R | |
| Panitumumab | ± Gemcitabine + Cisplatin | II | NCT02546934 | R | |
| ± Carboplatin + Paclitaxel | II | NCT02593175 | R | ||
| ± Carboplatin + Paclitaxel + Doxorubicin + Cyclophosphamide | II | NCT02876107 | R | ||
| SCT200 | - | II | NCT03692689 | R | |
| HER2 | Trastuzumab | ± Paclitaxel + Cyclophosphamide | II | NCT01750073 | R |
| VEGF | Bevacizumab | ± Paclitaxel + Erlotinib (EGFRi) | II | NCT00733408 | Ac/NR |
| VEGFR2 | Apatinib | ± Fluzoparib (PARPi) | I | NCT03075462 | R |
| ± Vinorelbine | II | NCT03254654 | R | ||
| SRC and WNT signaling | |||||
| SRC | Dasatinib | - | II | NCT02720185 | R |
| WNT | LGK974 | ± PDR001 (anti-PD-1) | I | NCT01351103 | R |
| ± Gedatolisib (PI3Ki) | I | NCT03243331 | R | ||
| PI3K/AKT/mTOR pathway | |||||
| PIK3CA | Taselisib | ± Enzalutamide (ARi) | I/II | NCT02457910 | R |
| PIK3CB | IPI-549 | ± Nivolumab (anti PD-1) | I | NCT02637531 | R |
| PQR309 | ± Eribulin | I/II | NCT02723877 | R | |
| PI3K | AZD8186 | - | I | NCT01884285 | R |
| ± Docetaxel | I | NCT03218826 | R | ||
| BKM120 | ± Capecitabine + Trastuzumab (HER2i) | II | NCT02000882 | Ac/NR | |
| CUDC-907 | - | I | NCT02307240 | Ac/NR | |
| AKT | ARQ092 | ± Carboplatin + Paclitaxel | Ib | NCT02476955 | R |
| AZD5363 | ± Paclitaxel | II | NCT02423603 | Ac/NR | |
| GSK2141795 | ± Trametinib (MEKi) | II | NCT01964924 | Ac/NR | |
| Ipatasertib | - | II | NCT02162719 | Ac/NR | |
| ± Carboplatin + Paclitaxel | I/II | NCT03853707 | R | ||
| ± Paclitaxel | II/III | NCT03337724 | R | ||
| mTOR | AZD2014 | ± Selumetinib (ERKi) | I/II | NCT02583542 | Ac/NR |
| Everolimus | ± Cisplatin + Paclitaxel | I | NCT02120469 | Ac/NR | |
| ± Cisplatin + Paclitaxel | II | NCT02531932 | R | ||
| ± Doxorubicin + Bevacizumab (VEGFi) | II | NCT02456857 | R | ||
| Gedatolisib | ± Docetaxel + Cisplatin + Dacomitinib (EGFRi) | I | NCT01920061 | R | |
| ± Tak-117 (PI3Ki) + Cisplatin + Paclitaxel | II | NCT03193853 | R | ||
| Immune checkpoint | |||||
| PD-1 | Nivolumab | ± Capecitabine | II | NCT03487666 | R |
| ± Carboplatin | I | NCT03414684 | R | ||
| PDR001 | - | I/II | NCT02404441 | Ac/NR | |
| Pembrolizumab | - | I | NCT03197389 | R | |
| - | II | NCT02447003 | Ac/NR | ||
| - | II | NCT02644369 | Ac/NR | ||
| - | III | NCT02555657 | Ac/NR | ||
| ± Capecitabine ± Paclitaxel | I/II | NCT02734290 | R | ||
| ± Doxorubicin ± Aromatase inhibitors | II | NCT02648477 | R | ||
| ± Nab-paclitaxel (or Paclitaxel) + Carboplatin + Doxorubicin + Cyclophosphamide | I | NCT02622074 | Ac/NR | ||
| ± Nab-paclitaxel (or Paclitaxel) ± Gemcitabine + Carboplatin | III | NCT02819518, | Ac/NR | ||
| ± Paclitaxel + Carboplatin ± Doxorubicin ± Epirubicin + Cyclophosphamide | III | NCT03036488 | Ac/NR | ||
| ± Lenvatinib (TKI) | II | NCT03797326 | R | ||
| ± PVX-410 Vaccine | I | NCT03362060 | R | ||
| SHR1210 | ± Apatinib (TKI) | II | NCT03394287 | Ac/NR | |
| PD-L1 | Atezolizumab | ± Carboplatin | II | NCT03206203 | R |
| ± Carboplatin + Cyclophosphamide ± Paclitaxel | II | NCT01898117 | R | ||
| Durvalumab | ± Oleclumab + Paclitaxel + Carboplatin | I/II | NCT03616886 | R | |
| ± Paclitaxel | I/II | NCT02628132 | R | ||
| - | II | NCT02685059 | Ac/NR | ||
| FAZ053 | ± PDR001 | I | NCT02936102 | Ac/NR | |
| Target | Major Drug | Combinational Drug | Indication | Phase | NCT Identifiers | Status # |
|---|---|---|---|---|---|---|
| Receptor | ||||||
| IGF-1R | AMG479 (Ganitumab) | ± Dasatinb | Embryonal and alveolar rhabdomyo-sarcoma | I/II | NCT-03041701 | R |
| ± (Everolimus + Panitumumab) | Advanced solid tumors, NSCLC | I | NCT-01061788 | Ac/NR | ||
| ± Vincristine + Doxorubicin + Cyclo-phosphamide / Ifosfamide + Etoposide | Metastatic malignant neoplasm in the bone, bone marrow, lung, and etc. | III | NCT-02306161 | Ac/NR | ||
| Cixutumumab | ± Lapatinib ditosylate + Capecitabine | Breast cancer | II | NCT-00684983 | Ac/NR | |
| ± Paclitaxel | Esophageal cancer, Gastro-esophageal junction adeno-carcinoma | II | NCT-01142388 | Ac/NR | ||
| DNA Plasmid Based Vaccine (WOKVAC) | - | Breast cancer | I | NCT-02780401 | R | |
| MK-0646 (Dalotuzumab) | ± Gemcitabine + Erlotinib | Advanced pancreatic cancer | I/II | NCT-00769483 | Ac/NR | |
| Teprotumumab | - | Thyroid eye disease, Graves’ orbitopathy | III | NCT-03298867 | Ac/NR | |
| Trastuzumab | ± Lapatinib ditosylate | Breast cancer | III | NCT-01104571 | Ac/NR | |
| nAChR | Bupropion | - | Breast cancer | III | NCT-03996265 | R |
| Cytokine | ||||||
| IL-6 receptor | Tocilizumab | ± Ipilimumab + Nivolumab | Advanced melanoma | II | NCT-03999749 | R |
| IL-7 receptor | Tocilizumab | ± Gemcitabine + nab-Paclitaxel | Unresectable pancreatic carcinoma | II | NCT-02767557 | R |
| Dormancy and proliferation | ||||||
| All-trans retinoic acid (ATRA) | ± 5-Azacitidine | Prostate cancer | II | NCT-03572387 | R | |
| Metabolism | ||||||
| Glycolysis inhibition | 2-Deoxyglucose (2DG) | - | Advanced cancer and hormone refractory prostate cancer | I/II | NCT-00633087 | T |
| Autophagy pathway | ||||||
| Autophagy inhibition | Hydro-chloroquine | - | Breast cancer | II | NCT-01292408 | U |
| Chloroquine | ± Carboplatin + Gemcitabine | Advanced solid tumors | I | NCT-02071537 | R | |
| + Temozolomide + Radiotherapy | Glioblastoma multiforme | I | NCT-02378532 | R | ||
| Drug resistance | ||||||
| Resistance inhibition | Doxycycline | - | Bone-targeted therapy in patients with metastatic breast cancer | II | NCT-01847976 | U |
| - | Cutaneous T-cell lymphoma | II | NCT-02341209 | R | ||
| Metformin | - | Colon cancer | II | NCT-03359681 | NR | |
| - | Prostate cancer | II | NCT-03137186 | R | ||
| - | Prostate cancer | II | NCT-02176161 | R | ||
| - | Breast cancer prevention | III | NCT-01905046 | R | ||
| + Doxycycline | Breast and uterine corpus cancer | II | NCT-02874430 | R | ||
| + Erlotinib | TNBC | I | NCT-01650506 | C | ||
| + Paclitaxel + Carboplatin + Docetaxel | Ovarian, primary peritoneal, or fallopian tube carcinoma | II | NCT-02122185 | R | ||
| + Pembrolizumab | Melanoma | I | NCT-03311308 | R | ||
| + Temozolomide | Glioblastoma | II | NCT-03243851 | |||
| + Temsirolimus | Advanced cancers | I | NCT-01529593 | Ac/NR | ||
| Others | ||||||
| CD44 | RO5429083 | - | CD44-expressing, malignant solid tumors | I | NCT-01358903 | C |
| ER | Tamoxifen | + 9-Cis-retinoic acid | Breast cancer | I | NCT-00001504 | C |
| GD2 | 3F8/GM-CSF | + 13-Cis-retinoic acid | Primary refractory neuroblastoma in bone marrow | II | NCT-01183897 | Ac/NR |
| Hedgehog | Vismodegib | - | Basal cell carcinoma | II | NCT-03035188 | R |
| + RO4929097 | Breast cancer | I | NCT-01071564 | T | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-L.; Kuo, Y.-C.; Ho, Y.-S.; Huang, Y.-H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers 2019, 11, 1334. https://doi.org/10.3390/cancers11091334
Lee K-L, Kuo Y-C, Ho Y-S, Huang Y-H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers. 2019; 11(9):1334. https://doi.org/10.3390/cancers11091334
Chicago/Turabian StyleLee, Kha-Liang, Yung-Che Kuo, Yuan-Soon Ho, and Yen-Hua Huang. 2019. "Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness" Cancers 11, no. 9: 1334. https://doi.org/10.3390/cancers11091334
APA StyleLee, K. -L., Kuo, Y. -C., Ho, Y. -S., & Huang, Y. -H. (2019). Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers, 11(9), 1334. https://doi.org/10.3390/cancers11091334