Targeting Autophagy by MPT0L145, a Highly Potent PIK3C3 Inhibitor, Provides Synergistic Interaction to Targeted or Chemotherapeutic Agents in Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
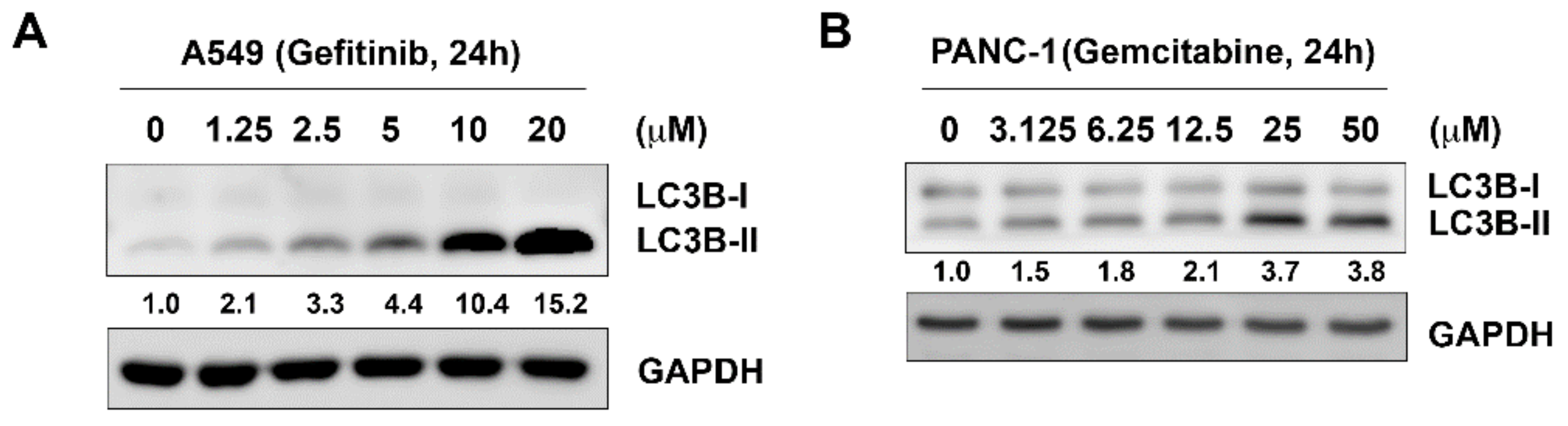
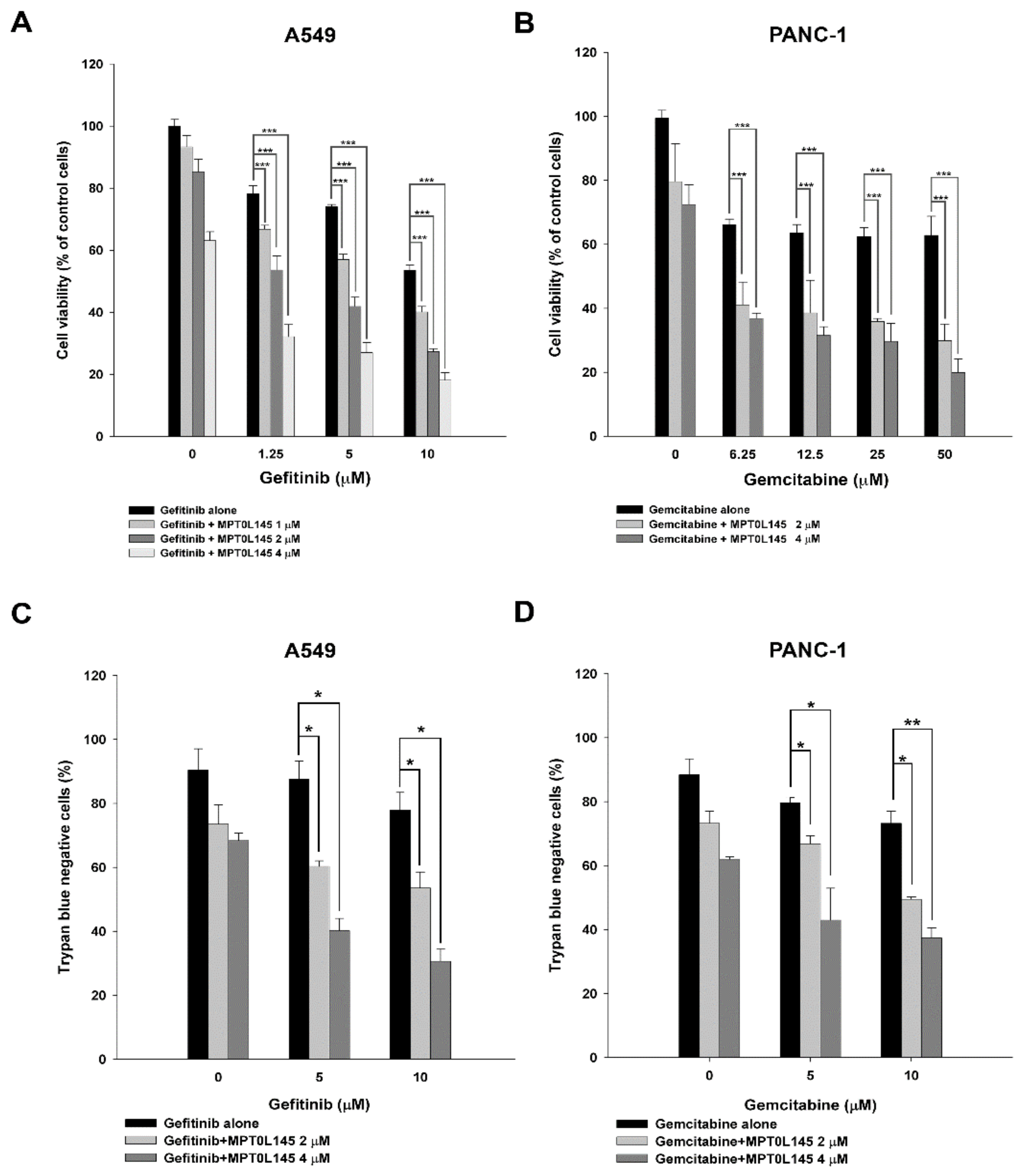
2.1. Combined-Use of MPT0L145 with Targeted or Chemotherapeutic Agents Provides Synthetic Lethality in Cancer Cells
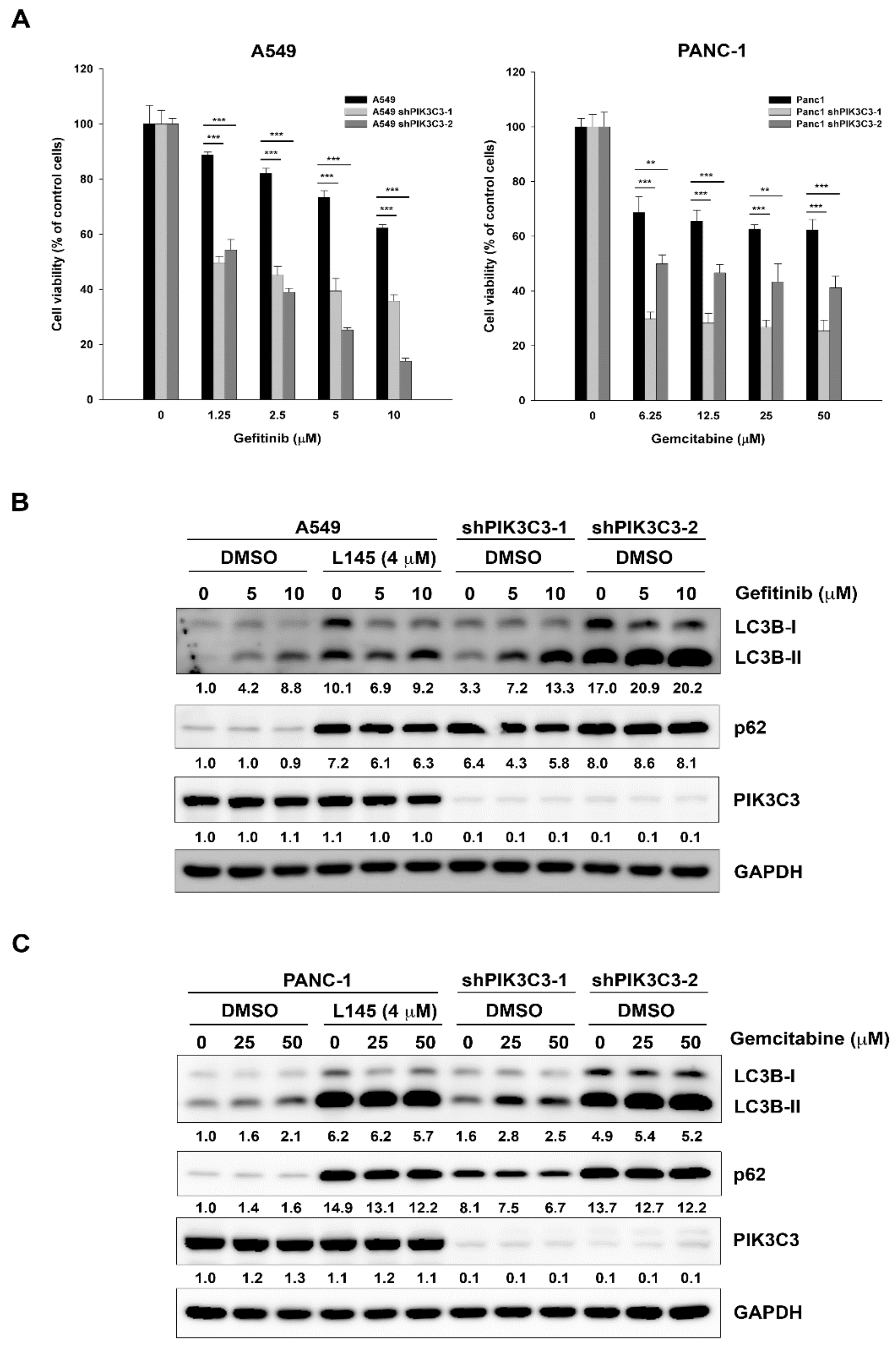
2.2. PIK3C3 Knockdown Mimics the Effects of MPT0L145
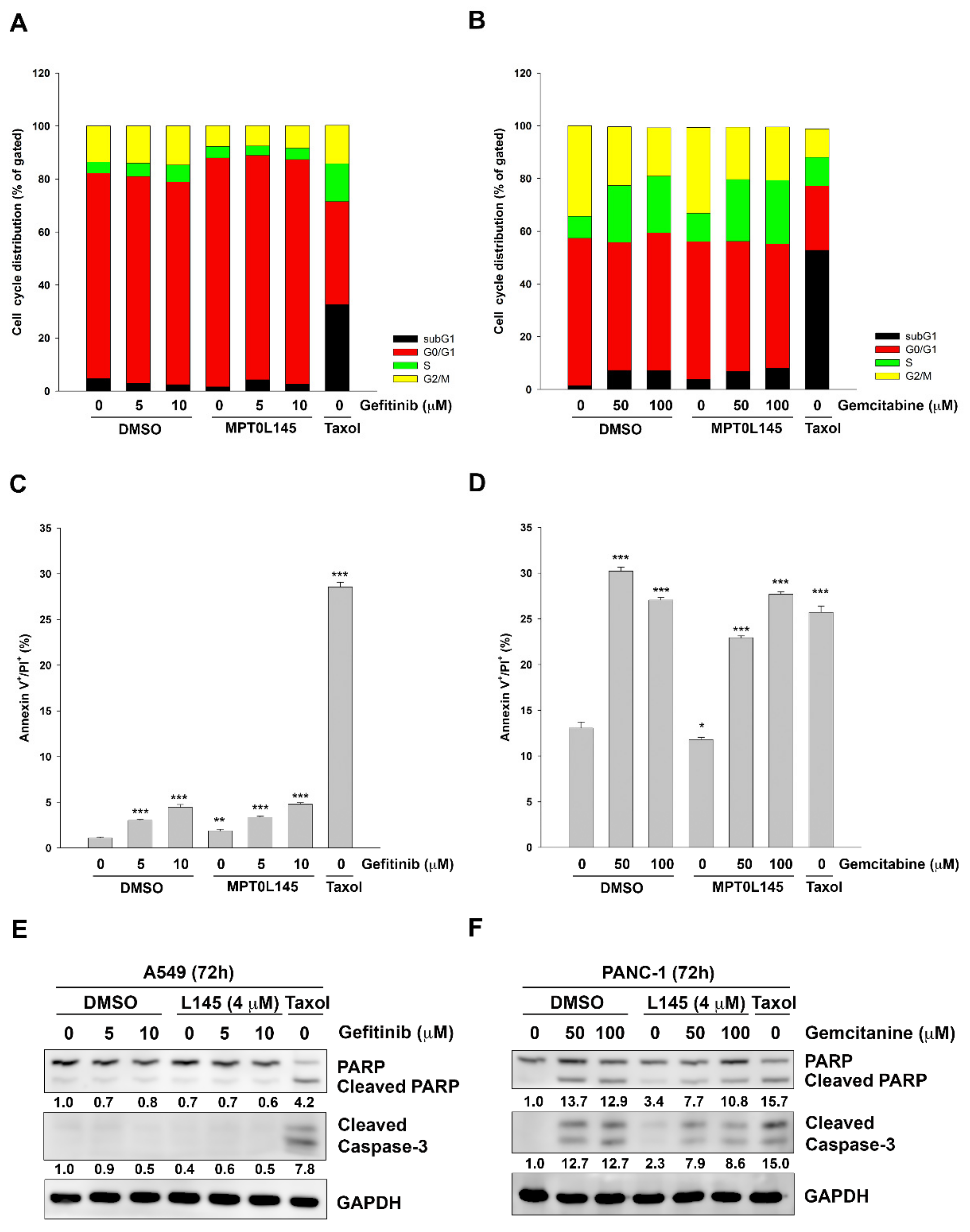
2.3. Drug Combination Displays no Effect on Cell Cycle and Apoptosis
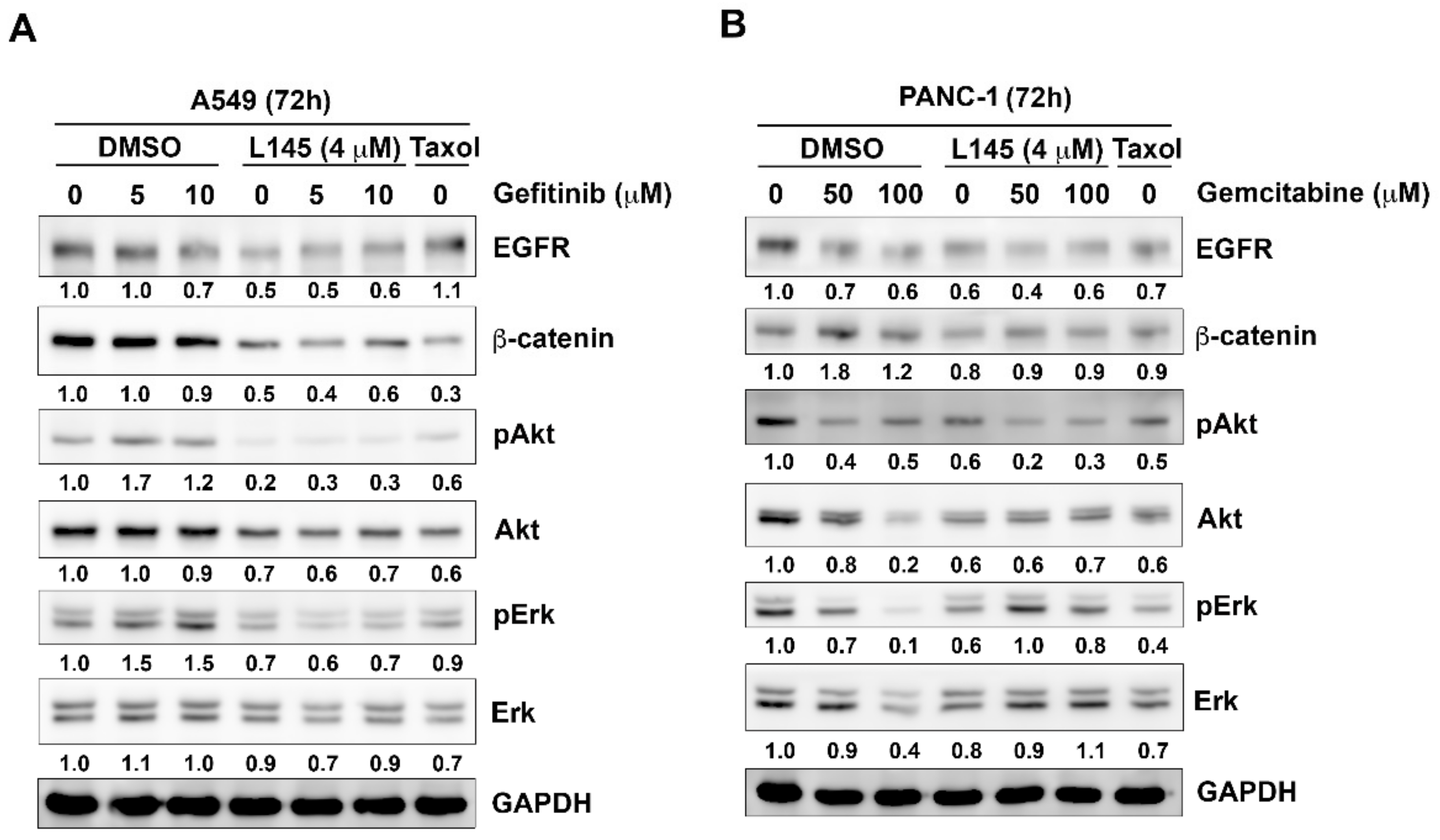
2.4. Drug Combination Perturbs Cell Survival Pathways in Cancer Cells
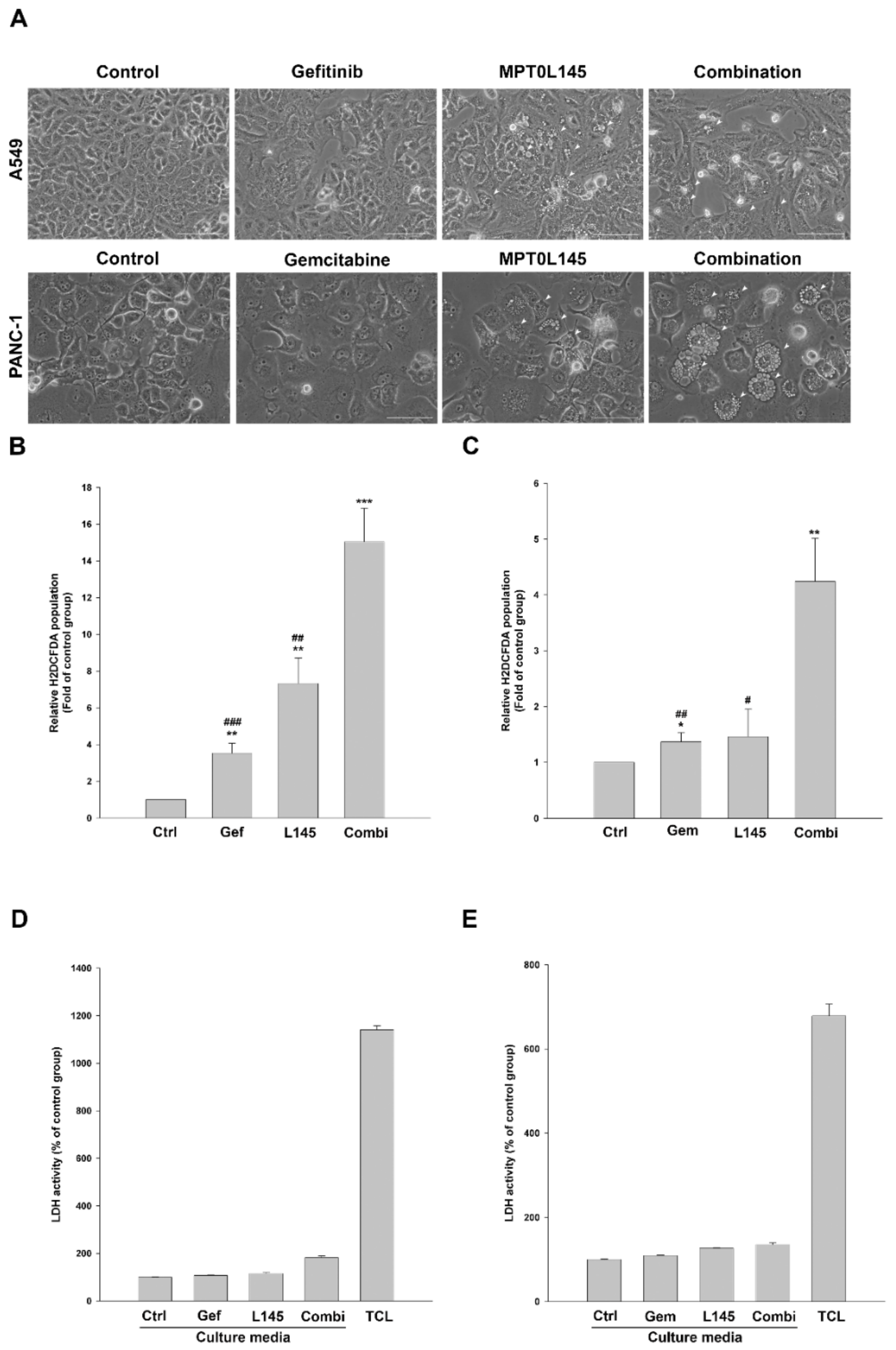
2.5. Drug Combination Increases Intracellular Vacuolization and Reactive Oxygen Species (ROS) Level
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Antibodies and Reagents
4.2. Cell Viability, Trypan Blue Exclusion Assay and Lactate Dehydrogenase (LDH) Assay
4.3. Cell Cycle and Apoptosis Analysis
4.4. Intracellular ROS Analysis
4.5. Western Blot Analysis and Lentiviral Expression System
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Carra, S.; Zhu, W.G.; Kampinga, H.H. The regulation of the autophagic network and its implications for human disease. Int. J. Biol. Sci. 2013, 9, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, U.K.; Chaudhary, A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 2015, 7, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Shi, T.T.; Yu, X.X.; Yan, L.J.; Xiao, H.T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 2017, 79, 287–294. [Google Scholar] [CrossRef]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine (Baltimore) 2018, 97, e12912. [Google Scholar] [CrossRef]
- Yusuf, I.H.; Sharma, S.; Luqmani, R.; Downes, S.M. Hydroxychloroquine retinopathy. Eye (Lond.) 2017, 31, 828–845. [Google Scholar] [CrossRef]
- Leung, L.S.; Neal, J.W.; Wakelee, H.A.; Sequist, L.V.; Marmor, M.F. Rapid Onset of Retinal Toxicity From High-Dose Hydroxychloroquine Given for Cancer Therapy. Am. J. Ophthalmol. 2015, 160, 799–805. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.H.; Yip, C.K.; Shi, Y.; Chait, B.T.; Wang, Q.J. Beclin 1-Vps34 Complex Architecture: Understanding the Nuts and Bolts of Therapeutic Targets. Front Biol. (Beijing) 2015, 10, 398–426. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex—At the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. Autophagy inhibitors. Cell Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Kim, J. Novel pharmacological modulators of autophagy: an updated patent review (2012-2015). Expert. Opin. Ther. Pat. 2016, 26, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Changou, C.A.; Hsieh, T.H.; Lee, Y.C.; Chu, C.Y.; Hsu, K.C.; Wang, H.C.; Lin, Y.C.; Lo, Y.N.; Liu, Y.R.; et al. Dual Inhibition of PIK3C3 and FGFR as a New Therapeutic Approach to Treat Bladder Cancer. Clin. Cancer Res. 2018, 24, 1176–1189. [Google Scholar] [CrossRef]
- Chen, C.H.; Liu, Y.M.; Pan, S.L.; Liu, Y.R.; Liou, J.P.; Yen, Y. Trichlorobenzene-substituted azaaryl compounds as novel FGFR inhibitors exhibiting potent antitumor activity in bladder cancer cells in vitro and in vivo. Oncotarget 2016, 7, 26374–26387. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef]
- Raufi, A.G.; Manji, G.A.; Chabot, J.A.; Bates, S.E. Neoadjuvant Treatment for Pancreatic Cancer. Semin. Oncol. 2019, 46, 19–27. [Google Scholar] [CrossRef]
- Tang, M.C.; Wu, M.Y.; Hwang, M.H.; Chang, Y.T.; Huang, H.J.; Lin, A.M.; Yang, J.C. Chloroquine enhances gefitinib cytotoxicity in gefitinib-resistant nonsmall cell lung cancer cells. PLoS ONE 2015, 10, e0119135. [Google Scholar] [CrossRef]
- Hashimoto, D.; Blauer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Hirata, A.; Kometani, T.; Miyagawa, M.; Ueda, S.; Kinoshita, H.; Fujii, T.; Kuwano, M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol. Cancer Ther. 2004, 3, 465–472. [Google Scholar] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers (Basel) 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Gu, P.; Zhou, C.; Liang, A.; Ren, S.; Liu, F.; Zeng, Y.; Wu, Y.; Zhao, Y.; Huang, B.; et al. beta-Catenin overexpression is associated with gefitinib resistance in non-small cell lung cancer cells. Pulm. Pharmacol. Ther. 2014, 28, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Nakata, A.; Yoshida, R.; Yamaguchi, R.; Yamauchi, M.; Tamada, Y.; Fujita, A.; Shimamura, T.; Imoto, S.; Higuchi, T.; Nomura, M.; et al. Elevated beta-catenin pathway as a novel target for patients with resistance to EGF receptor targeting drugs. Sci. Rep. 2015, 5, 13076. [Google Scholar] [CrossRef]
- Zheng, C.; Jiao, X.; Jiang, Y.; Sun, S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J. Int. Med. Res. 2013, 41, 300–306. [Google Scholar] [CrossRef]
- Chen, D.; Niu, M.; Jiao, X.; Zhang, K.; Liang, J.; Zhang, D. Inhibition of AKT2 enhances sensitivity to gemcitabine via regulating PUMA and NF-kappaB signaling pathway in human pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2012, 13, 1186–1208. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef]
- Bellizzi, J.J., 3rd; Widom, J.; Kemp, C.; Lu, J.Y.; Das, A.K.; Hofmann, S.L.; Clardy, J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc. Natl. Acad. Sci. USA 2000, 97, 4573–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Ling, Y.H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Ito, K.; Yamashiro, Y.; Moriya, S.; Che, X.F.; Yokoyama, T.; Hiramoto, M.; Miyazawa, K. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohue, E.; Thomas, A.; Maurer, N.; Manisali, I.; Zeisser-Labouebe, M.; Zisman, N.; Anderson, H.J.; Ng, S.S.; Webb, M.; Bally, M.; et al. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. J. Cancer 2013, 4, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yang, G.; Shi, Y.; Su, C.; Chang, J. Co-delivery of Gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. J. Nanobiotechnol. 2015, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Xu, N.; O’Prey, J.; Lao, L.Y.; Joshi, S.; Long, J.S.; O’Prey, M.; Croft, D.R.; Beaumatin, F.; Baudot, A.D.; et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad Sci. USA 2015, 112, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef]
- Wang, Z.; Du, T.; Dong, X.; Li, Z.; Wu, G.; Zhang, R. Autophagy inhibition facilitates erlotinib cytotoxicity in lung cancer cells through modulation of endoplasmic reticulum stress. Int. J. Oncol. 2016, 48, 2558–2566. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. Autophagy inhibition improves anti-cancer drugs via FOXO3a activation. Oncotarget 2018, 9, 25384–25385. [Google Scholar] [CrossRef]
- Han, H.; Chou, C.C.; Li, R.; Liu, J.; Zhang, L.; Zhu, W.; Hu, J.; Yang, B.; Tian, J. Chalcomoracin is a potent anticancer agent acting through triggering Oxidative stress via a mitophagy- and paraptosis-dependent mechanism. Sci. Rep. 2018, 8, 9566. [Google Scholar] [CrossRef]
- Lee, W.J.; Chien, M.H.; Chow, J.M.; Chang, J.L.; Wen, Y.C.; Lin, Y.W.; Cheng, C.W.; Lai, G.M.; Hsiao, M.; Lee, L.M. Nonautophagic cytoplasmic vacuolation death induction in human PC-3M prostate cancer by curcumin through reactive oxygen species -mediated endoplasmic reticulum stress. Sci. Rep. 2015, 5, 10420. [Google Scholar] [CrossRef]
- Li, Z.; Mbah, N.E.; Overmeyer, J.H.; Sarver, J.G.; George, S.; Trabbic, C.J.; Erhardt, P.W.; Maltese, W.A. The JNK signaling pathway plays a key role in methuosis (non-apoptotic cell death) induced by MOMIPP in glioblastoma. BMC Cancer 2019, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef] [PubMed]
- Ardito, C.M.; Gruner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Martinelli, E.; Capasso, A.; Morgillo, F.; Orditura, M.; De Vita, F.; Ciardiello, F. Targeting EGFR in pancreatic cancer treatment. Curr. Drug Targets 2012, 13, 802–810. [Google Scholar] [CrossRef]
- Richard, C.; Niogret, J.; Boidot, R.; Ghiringhelli, F. EGFR amplification induces sensitivity to antiEGFR therapy in pancreatic acinar cell carcinoma. World J. Gastrointest. Oncol. 2018, 10, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Yuan, J.Q.; Di, M.Y.; Zheng, D.Y.; Chen, J.Z.; Ding, H.; Wu, X.Y.; Huang, Y.F.; Mao, C.; Tang, J.L. Gemcitabine plus erlotinib for advanced pancreatic cancer: a systematic review with meta-analysis. PLoS ONE 2013, 8, e57528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, X.K.; Wang, W.X.; Zhang, R.M.; Zhang, T.; Ren, J.J. The Wnt/beta-catenin signaling pathway mechanism for pancreatic cancer chemoresistance in a three-dimensional cancer microenvironment. Am. J. Transl. Res. 2016, 8, 4490–4498. [Google Scholar]
- Jia, Y.; Xie, J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.M.; Chen, C.H.; Yeh, T.K.; Liou, J.P. Synthesis and evaluation of novel 7H-pyrrolo-[2,3-d]pyrimidine derivatives as potential anticancer agents. Future Med. Chem. 2019. [Google Scholar] [CrossRef]
- Romani, M.; Daga, A.; Forlani, A.; Pistillo, M.P.; Banelli, B. Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells. Cancers (Basel) 2019, 11, 878. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-H.; Hsieh, T.-H.; Lin, Y.-C.; Liu, Y.-R.; Liou, J.-P.; Yen, Y. Targeting Autophagy by MPT0L145, a Highly Potent PIK3C3 Inhibitor, Provides Synergistic Interaction to Targeted or Chemotherapeutic Agents in Cancer Cells. Cancers 2019, 11, 1345. https://doi.org/10.3390/cancers11091345
Chen C-H, Hsieh T-H, Lin Y-C, Liu Y-R, Liou J-P, Yen Y. Targeting Autophagy by MPT0L145, a Highly Potent PIK3C3 Inhibitor, Provides Synergistic Interaction to Targeted or Chemotherapeutic Agents in Cancer Cells. Cancers. 2019; 11(9):1345. https://doi.org/10.3390/cancers11091345
Chicago/Turabian StyleChen, Chun-Han, Tsung-Han Hsieh, Yu-Chen Lin, Yun-Ru Liu, Jing-Ping Liou, and Yun Yen. 2019. "Targeting Autophagy by MPT0L145, a Highly Potent PIK3C3 Inhibitor, Provides Synergistic Interaction to Targeted or Chemotherapeutic Agents in Cancer Cells" Cancers 11, no. 9: 1345. https://doi.org/10.3390/cancers11091345
APA StyleChen, C. -H., Hsieh, T. -H., Lin, Y. -C., Liu, Y. -R., Liou, J. -P., & Yen, Y. (2019). Targeting Autophagy by MPT0L145, a Highly Potent PIK3C3 Inhibitor, Provides Synergistic Interaction to Targeted or Chemotherapeutic Agents in Cancer Cells. Cancers, 11(9), 1345. https://doi.org/10.3390/cancers11091345