Whole Genome Sequencing of Spontaneously Occurring Rat Natural Killer Large Granular Lymphocyte Leukemia Identifies JAK1 Somatic Activating Mutation


 , ,
, ,
Abstract
:1. Introduction
2. Results
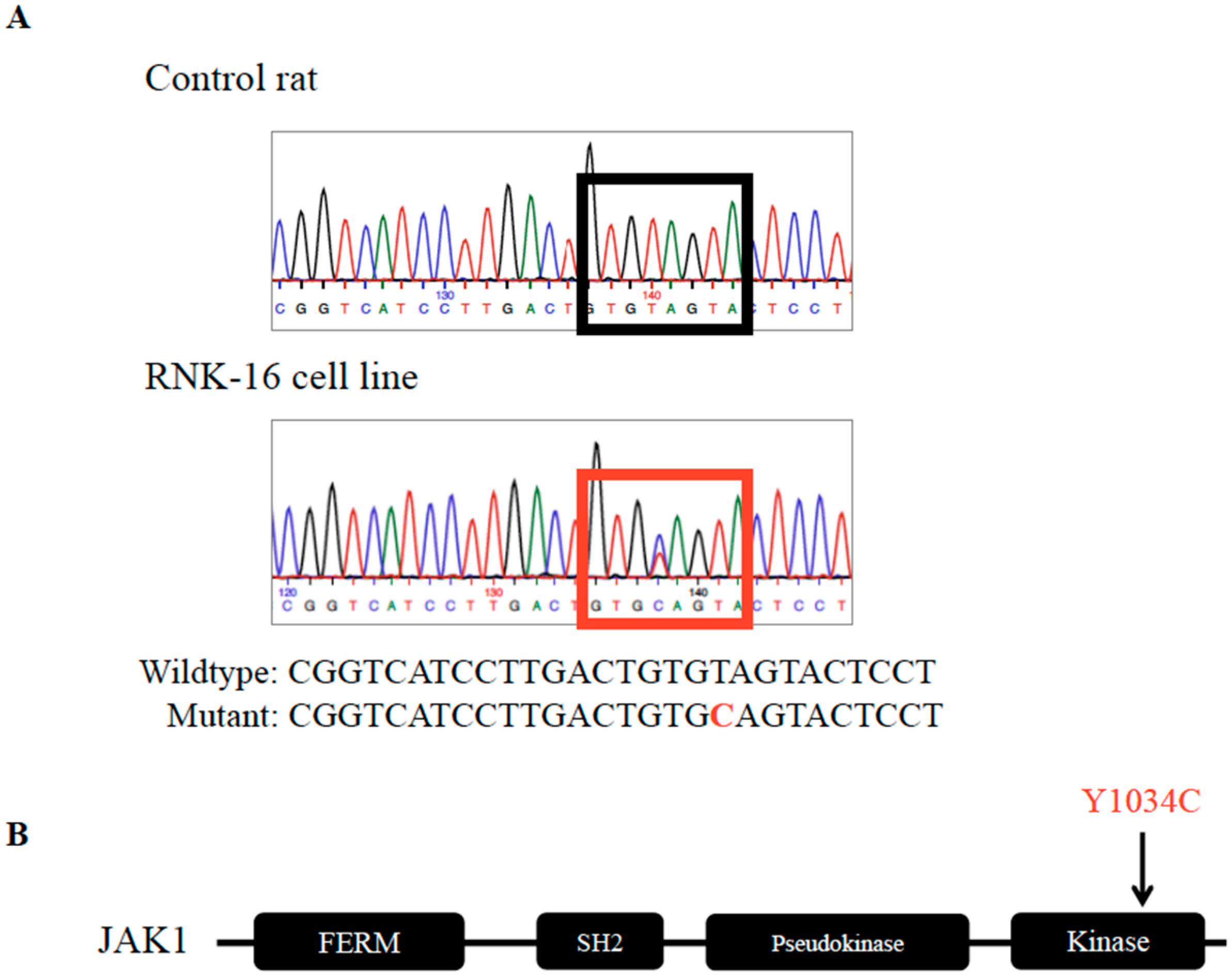
2.1. Identification and Validation of Mutations in the RNK-16 Cell Line and RNK-16 Primary Spleen Material
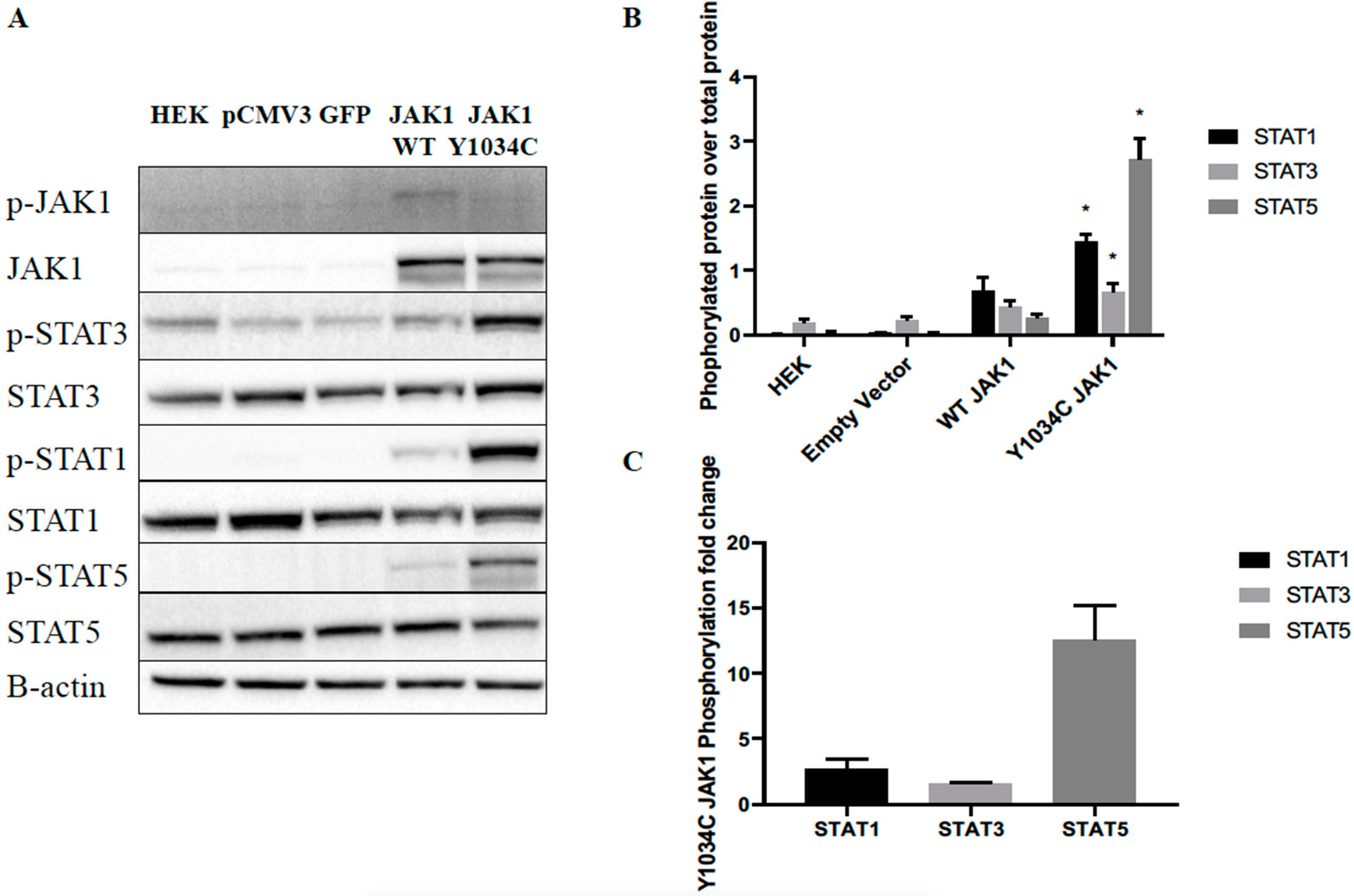
2.2. JAK1 Mutation Increased Downstream STAT Signaling
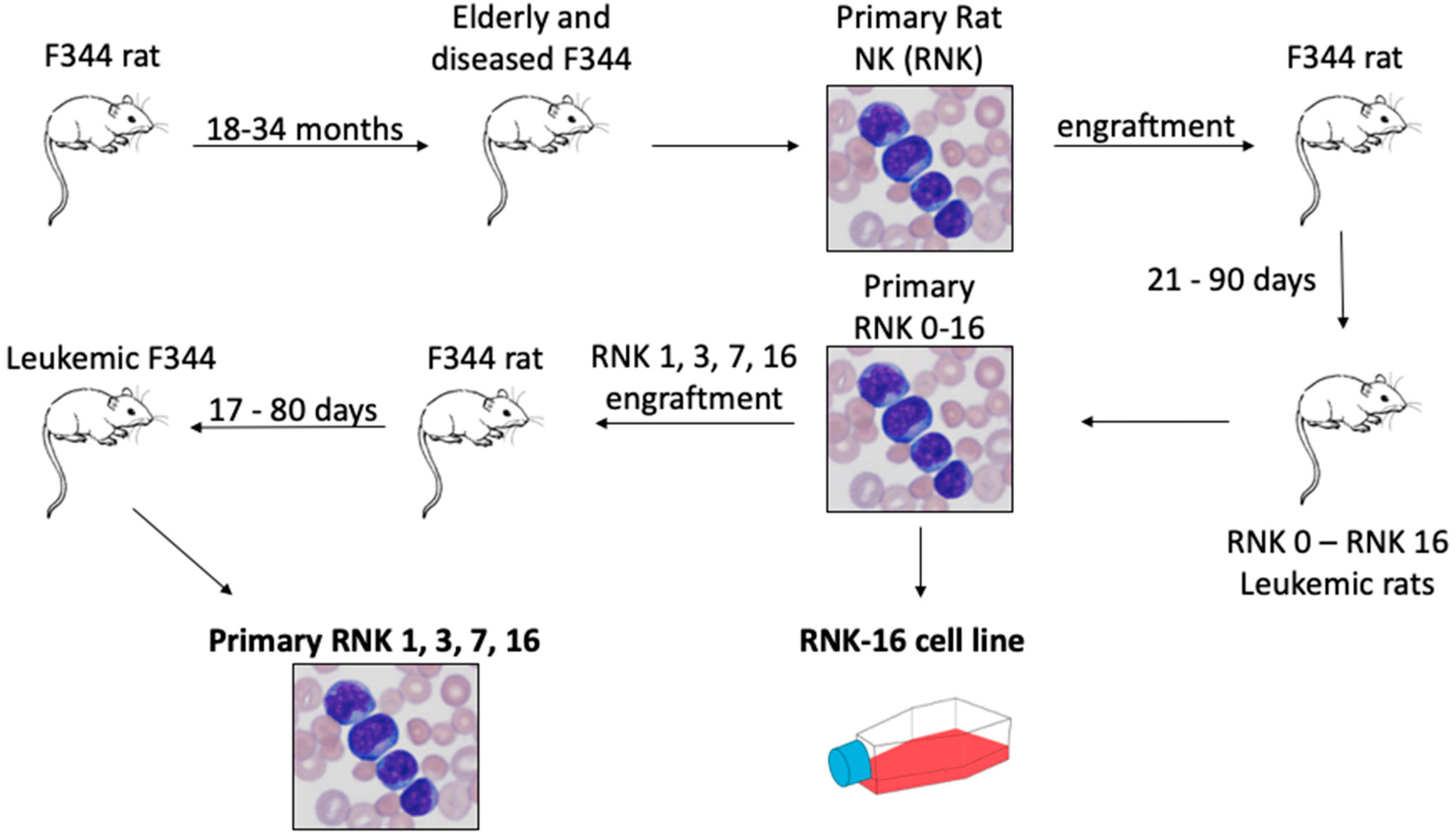
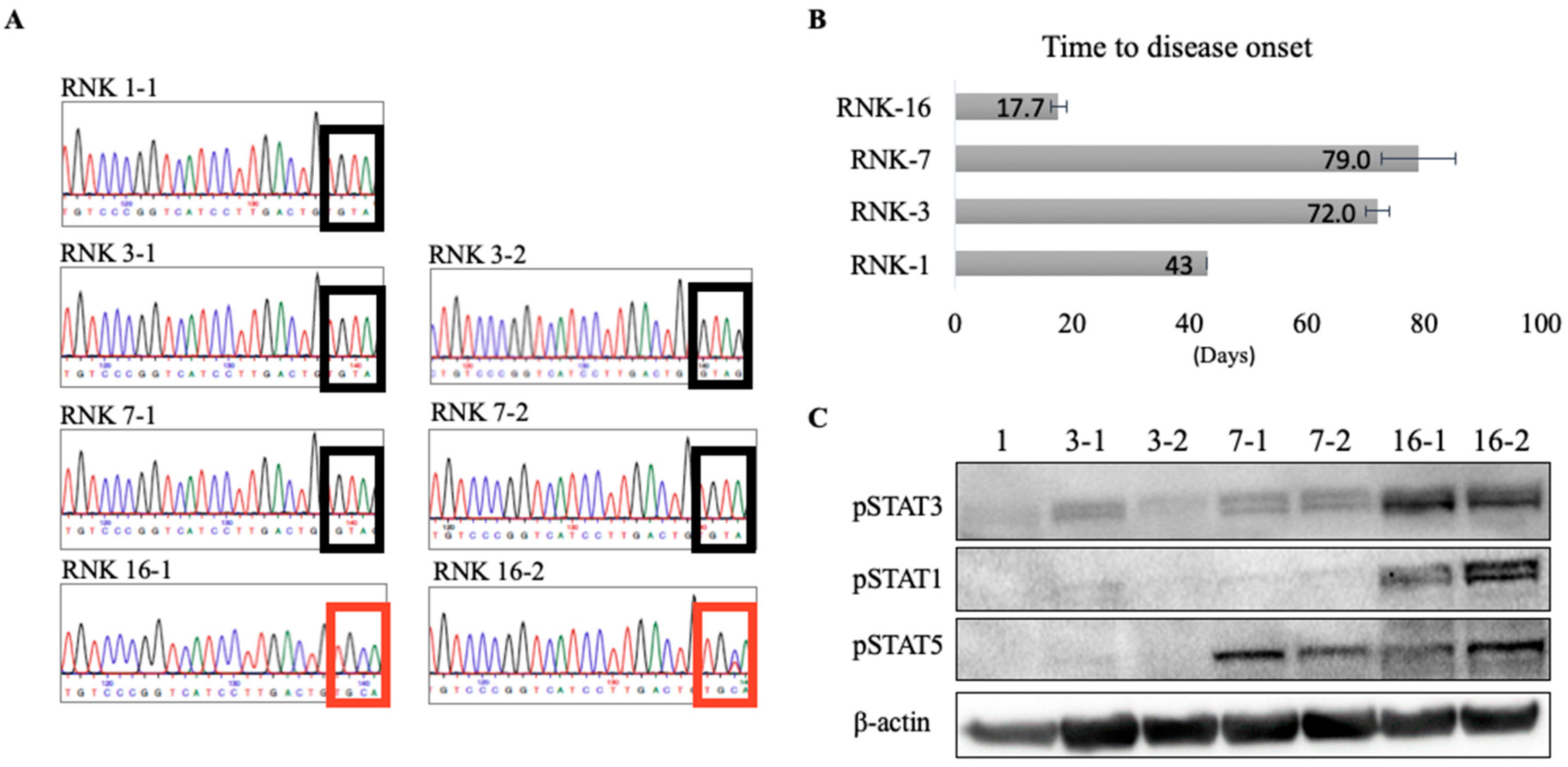
2.3. In Vivo and Ex Vivo Characterization of Primary Rat Natural Killer Material Shows an Aggressive Disease Course and Increased STAT Signaling With Mutant JAK1
3. Discussion
4. Materials and Methods
4.1. Acquisition of Sequencing Data
4.2. Alignments
4.3. CNV Map
4.4. Identification of Variants
4.5. Annotation and Filtering of Variants
4.6. Gene Enrichment Analyses
4.7. Sanger Sequencing to Validate Whole Genome Sequencing Results
4.8. JAK1 Mutagenesis
4.9. Cell Culture
4.10. Transfection and Western Blot Analysis
4.11. Animal Study
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watters, R.J.; Liu, X.; Loughran, T.P. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk. Lymphoma 2011, 52, 2217–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, R.; Suzumiya, J.; Nakamura, S.; Aoki, S.; Notoya, A.; Ozaki, S.; Gondo, H.; Hino, N.; Mori, H.; Sugimori, H.; et al. Aggressive natural killer-cell leukemia revisited: Large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia 2004, 18, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamy, T.; Moignet, A.; Loughran, T.P. LGL leukemia: From pathogenesis to treatment. Blood 2017, 129, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Leblanc, F.; Loughran, T.P. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev. 2014, 28, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Ward, A.C.; Touw, I.; Yoshimura, A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 2000, 95, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, B.; Hu, L.; Ning, H.; Jiang, M.; Wang, D.; Liu, T.; Zhang, B.; Chen, H. Identification of a novel functional JAK1 S646P mutation in acute lymphoblastic leukemia. Oncotarget 2017, 8, 34687–34697. [Google Scholar] [CrossRef] [Green Version]
- Flex, E.; Petrangeli, V.; Stella, L.; Chiaretti, S.; Hornakova, T.; Knoops, L.; Ariola, C.; Fodale, V.; Clappier, E.; Paoloni, F.; et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 2008, 205, 751–758. [Google Scholar] [CrossRef]
- Jeong, E.G.; Kim, M.S.; Nam, H.K.; Min, C.K.; Lee, S.; Chung, Y.J.; Yoo, N.J.; Lee, S.H. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin. Cancer Res. 2008, 14, 3716–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanger, D.; Jacquemin, V.; Chopin, M.; Pierron, G.; Bernard, O.A.; Ghysdael, J.; Stern, M.-H. Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia 2014, 28, 417–419. [Google Scholar] [CrossRef]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Zhao, Y.; Mitaksov, V.; Fremont, D.H.; Kasai, Y.; Molitoris, A.; Ries, R.E.; Miner, T.L.; McLellan, M.D.; DiPersio, J.F.; et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood 2008, 111, 4809–4812. [Google Scholar] [CrossRef] [Green Version]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, H.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhang, Y.; Liu, R.Z.; Fenstermacher, D.A.; Wright, K.L.; Teer, J.K.; Wu, J. JAK1 truncating mutations in gynecologic cancer define new role of cancer-associated protein tyrosine kinase aberrations. Sci. Rep. 2013, 3, 3042. [Google Scholar] [CrossRef] [Green Version]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.-M.; Yang-Yen, H.-F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.C.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, J.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun 2015, 6, 6025. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.I.; Tanahashi, T.; Sekiguchi, N.; Gasparini, V.R.; Bortoluzzi, S.; Kawakami, T.; Matsuda, K.; Mitsui, T.; Eldfors, S.; Bortoluzzi, S.; et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood 2016, 128, 2465–2468. [Google Scholar] [CrossRef] [Green Version]
- Rajala, H.L.M.; Porkka, K.; Maciejewski, J.P.; Loughran, T.P.; Mustjoki, S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann. Med. 2014, 46, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.L.M.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.; Kuusanmäki, H.; Bortoluzzi, S.; Lagström, S.; Parsons, A.; Rajala, H.; van Adrichem, A.; Eldfors, S.; Olson, T.; Clemente, M.J.; et al. Activating somatic mutations outside the SH2-domain of STAT3 in LGL leukemia. Leukemia 2016, 30, 1204–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maronpot, R.R.; Nyska, A.; Foreman, J.E.; Ramot, Y. The legacy of the F344 rat as a cancer bioassay model (a retrospective summary of three common F344 rat neoplasms). Crit. Rev. Toxicol. 2016, 46, 641–675. [Google Scholar] [CrossRef]
- Stromberg, P.C. Large granular lymphocyte leukemia in F344 rats. Model for human T gamma lymphoma, malignant histiocytosis, and T-cell chronic lymphocytic leukemia. Am. J. Pathol. 1985, 119, 517–519. [Google Scholar]
- Reynolds, C.W.; Timonen, T.; Herberman, R.B. Natural killer (NK) cell activity in the rat. I. Isolation and characterization of the effector cells. J. Immunol. 1981, 127, 282–287. [Google Scholar]
- Losco, P.E.; Ward, J.M. The Early Stage of Large Granular Lymphocyte Leukemia in the F344 Rat. Vet. Pathol. 1984, 21, 286–291. [Google Scholar] [CrossRef]
- Thomas, J.; Haseman, J.K.; Goodman, J.I.; Ward, J.M.; Loughran, T.P.; Spencer, P.J. A review of large granular lymphocytic leukemia in fischer 344 Rats as an initial step toward evaluating the implication of the endpoint to human cancer risk assessment. Toxicol. Sci. 2007, 99, 3–19. [Google Scholar] [CrossRef]
- Ward, J.M.; Reynolds, C.W. Large granular lymphocyte leukemia. A heterogeneous lymphocytic leukemia in F344 rats. Am. J. Pathol. 1983, 111, 1–10. [Google Scholar]
- Reynolds, C.W.; Bere, E.W.; Ward, J.M. Natural killer activity in the rat. III. Characterization of transplantable large granular lymphocyte (LGL) leukemias in the F344 rat. J. Immunol. 1984, 132, 534–540. [Google Scholar]
- Ryan, J.C.; Niemi, E.C.; Nakamura, M.C. Functional analysis of natural killer cell receptors in the RNK-16 rat leukemic cell line. Methods Mol. Biol. 2000, 121, 283–295. [Google Scholar] [PubMed]
- Axberg, I.; Nose, M.; Reynolds, C.W.; Wigzell, H. Features of the in vitro established rat large granular lymphocyte leukaemia RNK-16. Scand. J. Immunol. 1988, 27, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.W.; Foon, K.A. T gamma-lymphoproliferative disease and related disorders in humans and experimental animals: A review of the clinical, cellular, and functional characteristics. Blood 1984, 64, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Ishmael, J.; Dugard, P.H. A review of perchloroethylene and rat mononuclear cell leukemia. Regul. Toxicol. Pharmacol. 2006, 45, 178–184. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Kim, M.; Rhee, J.-K.; Choi, H.; Kwon, A.; Kim, J.; Lee, G.D.; Jekarl, D.W.; Lee, S.; Kim, Y.; Kim, T.-M.; et al. Passage-Dependent Accumulation Of Somatic Mutations In Mesenchymal Stromal Cells During In Vitro Culture Revealed By Whole Genome Sequencing. Sci. Rep. 2017, 7, 14508. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5674020/ (accessed on 12 September 2019). [CrossRef] [Green Version]
- Coppe, A.; Andersson, E.I.; Binatti, A.; Gasparini, V.R.; Bortoluzzi, S.; Clemente, M.; Herling, M.; Maciejewski, J.; Mustjoki, S.; Bortoluzzi, S. Genomic landscape characterization of large granular lymphocyte leukemia with a systems genetics approach. Leukemia 2017, 31, 1243–1246. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.I.; Rajala, H.L.M.; Eldfors, S.; Ellonen, P.; Olson, T.; Jerez, A.; Clemente, M.J.; Kallioniemi, O.; Porkka, K.; Heckman, C.; et al. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J. 2013, 3, e168. [Google Scholar] [CrossRef] [Green Version]
- Klein, K.; Witalisz-Siepracka, A.; Maurer, B.; Prinz, D.; Heller, G.; Leidenfrost, N.; Prchal-Murphy, M.; Suske, T.; Moriggl, R.; Sexl, V.; et al. STAT5B N642H drives transformation of NKT cells: A novel mouse model for CD56 + T-LGL leukemia. Leukemia 2019, 33, 2336–2340. [Google Scholar] [CrossRef] [Green Version]
- Arulogun, S.O.; Choong, H.-L.; Taylor, D.; Ambrosoli, P.; Magor, G.; Irving, I.M.; Tee-Beng Keng, T.-P.; Perkins, A.C. JAK1 somatic mutation in a myeloproliferative neoplasm. Haematologica 2017, 102, e324–e327. [Google Scholar] [CrossRef] [Green Version]
- Gordon, G.M.; Lambert, Q.T.; Daniel, K.G.; Reuther, G.W. Transforming JAK1 mutations exhibit differential signalling, FERM domain requirements and growth responses to interferon-γ. Biochem. J. 2010, 432, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.D.; Gaffen, S.L.; Goldsmith, M.A.; Greene, W.C. Janus kinases in interleukin-2-mediated signaling: JAK1 and JAK3 are differentially regulated by tyrosine phosphorylation. Curr. Biol. 1997, 7, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Haan, S.; Margue, C.; Engrand, A.; Rolvering, C.; de Leur, H.S.-V.; Heinrich, P.C.; Behrmann, I.; Haan, V. Dual role of the Jak1 FERM and kinase domains in cytokine receptor binding and in stimulation-dependent Jak Activation. J. Immunol. 2008, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witalisz-Siepracka, A.; Klein, K.; Prinz, D.; Leidenfrost, N.; Schabbauer, G.; Dohnal, A.; Sexl, V. Loss of JAK1 drives innate immune deficiency. Front. Immunol. 2018, 9, 3108. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:13033997. Available online: http://arxiv.org/abs/1303.3997 (accessed on 10 September 2019).
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:12073907. Available online: http://arxiv.org/abs/1207.3907 (accessed on 3 October 2019).
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermsen, R.; de Ligt, J.; Spee, W.; Blokzijl, F.; Schäfer, S.; Adami, E.; Boymans, S.; Flink, S.; van Boxtel, R.; van der Weide, R.H.; et al. Genomic landscape of rat strain and substrain variation. BMC Genom. 2015, 16, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Tier Level | RNK-16 Cell Line WGS | RNK-16 Cell Line Validation | RNK-16 Primary Spleen Cells |
|---|---|---|---|---|
| Brd3 | 1 | ✓ | x | x |
| Ddr2 | 1 | ✓ | ✓ | x |
| Fat1 | 1 | ✓ | ✓ | x |
| Jak1 | 1 | ✓ | ✓ | ✓ |
| Ncor2 | 1 | ✓ | ✓ | x |
| Nono | 1 | ✓ | x | x |
| Stag | 1 | ✓ | ✓ | x |
| Thrap3 | 1 | ✓ | x | x |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.T.; Yang, J.; Dighe, S.; Schmachtenberg, M.W.; Leigh, N.T.; Farber, E.; Onengut-Gumuscu, S.; Feith, D.J.; Ratan, A.; Loughran, T.P., Jr.; et al. Whole Genome Sequencing of Spontaneously Occurring Rat Natural Killer Large Granular Lymphocyte Leukemia Identifies JAK1 Somatic Activating Mutation. Cancers 2020, 12, 126. https://doi.org/10.3390/cancers12010126
Wang TT, Yang J, Dighe S, Schmachtenberg MW, Leigh NT, Farber E, Onengut-Gumuscu S, Feith DJ, Ratan A, Loughran TP Jr., et al. Whole Genome Sequencing of Spontaneously Occurring Rat Natural Killer Large Granular Lymphocyte Leukemia Identifies JAK1 Somatic Activating Mutation. Cancers. 2020; 12(1):126. https://doi.org/10.3390/cancers12010126
Chicago/Turabian StyleWang, T. Tiffany, Jun Yang, Shubha Dighe, Matthew W. Schmachtenberg, Nathan T. Leigh, Emily Farber, Suna Onengut-Gumuscu, David J. Feith, Aakrosh Ratan, Thomas P. Loughran, Jr., and et al. 2020. "Whole Genome Sequencing of Spontaneously Occurring Rat Natural Killer Large Granular Lymphocyte Leukemia Identifies JAK1 Somatic Activating Mutation" Cancers 12, no. 1: 126. https://doi.org/10.3390/cancers12010126
APA StyleWang, T. T., Yang, J., Dighe, S., Schmachtenberg, M. W., Leigh, N. T., Farber, E., Onengut-Gumuscu, S., Feith, D. J., Ratan, A., Loughran, T. P., Jr., & Olson, T. L. (2020). Whole Genome Sequencing of Spontaneously Occurring Rat Natural Killer Large Granular Lymphocyte Leukemia Identifies JAK1 Somatic Activating Mutation. Cancers, 12(1), 126. https://doi.org/10.3390/cancers12010126