Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells


 and
and
Abstract
:1. Background
2. Methods and Materials
2.1. Cell Lines
2.2. Reagents
2.3. Proliferation Assay
2.4. Immunoblotting
2.5. P2Y12 Knockdown
2.6. ADP Secretion Assay
2.7. Apoptosis Assay
2.8. In Vivo Tumour Growth
2.9. Transforming Growth Factor Beta 1 (TGF-β1) ELISA
2.10. Statistical Analysis
2.11. Ethics Approval
3. Results
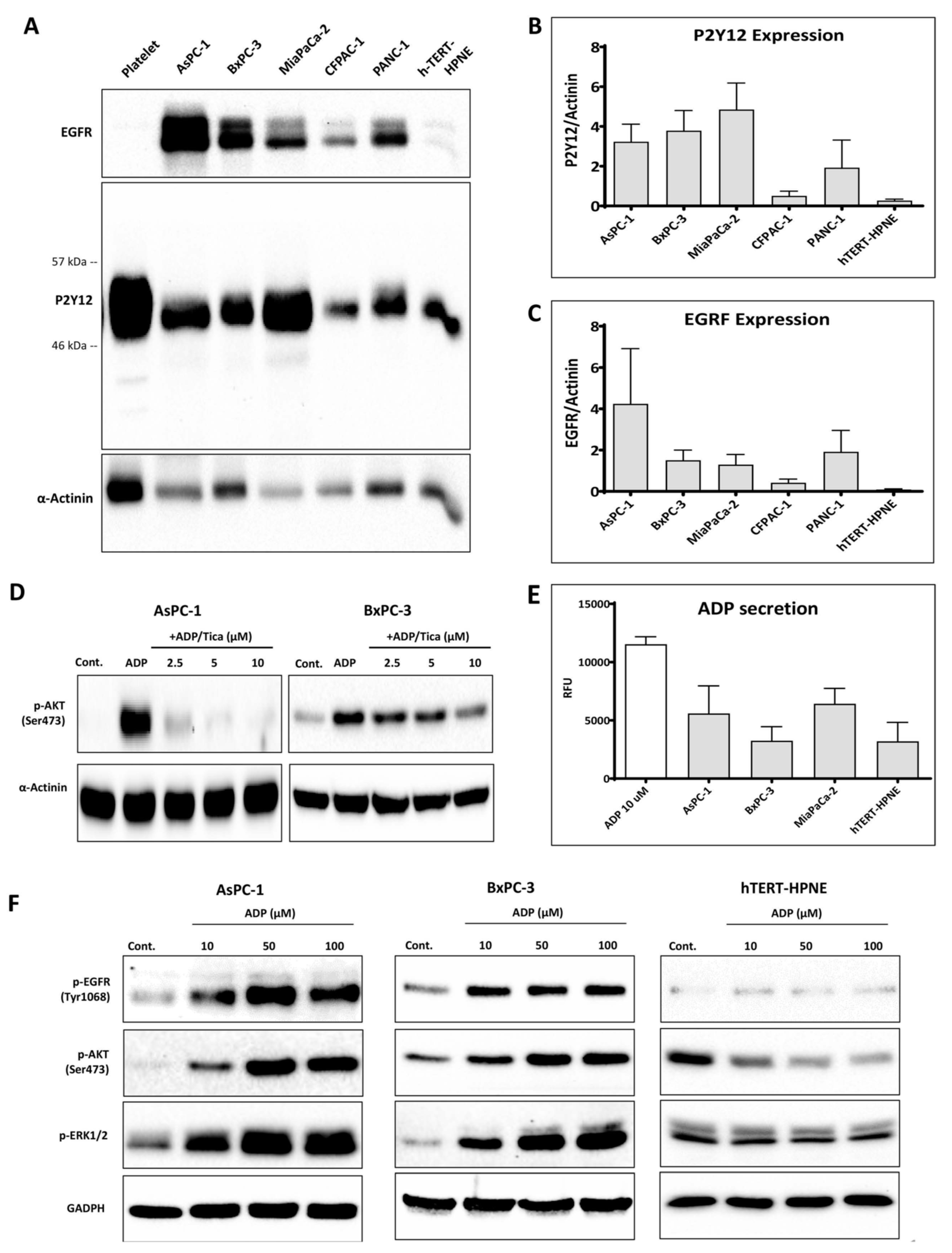
3.1. P2Y12 Expression in PDAC Cells
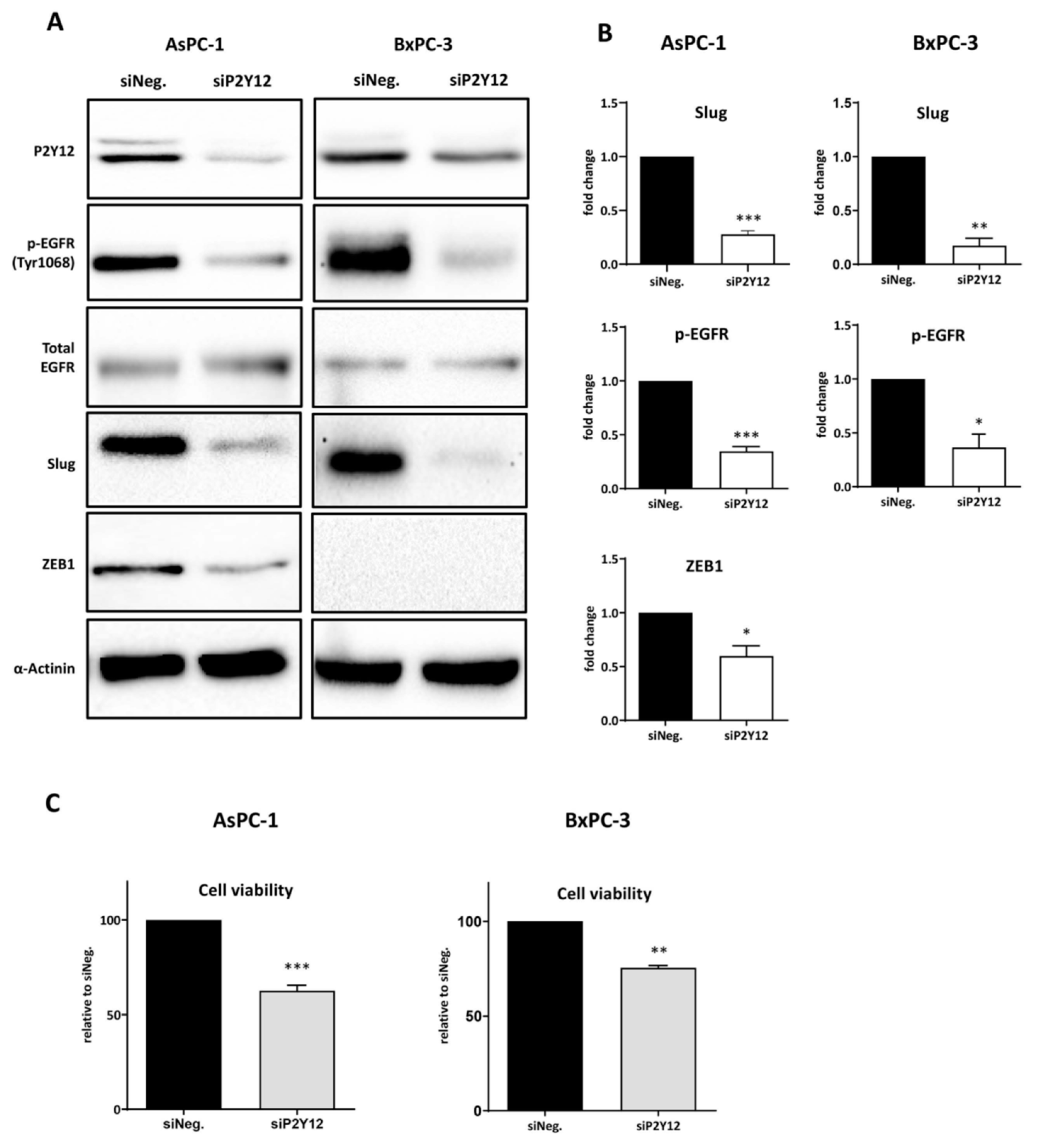
3.2. P2Y12 Regulated Phospho-EGFR, PDAC Cell Proliferation and EMT Markers SLUG and ZEB1
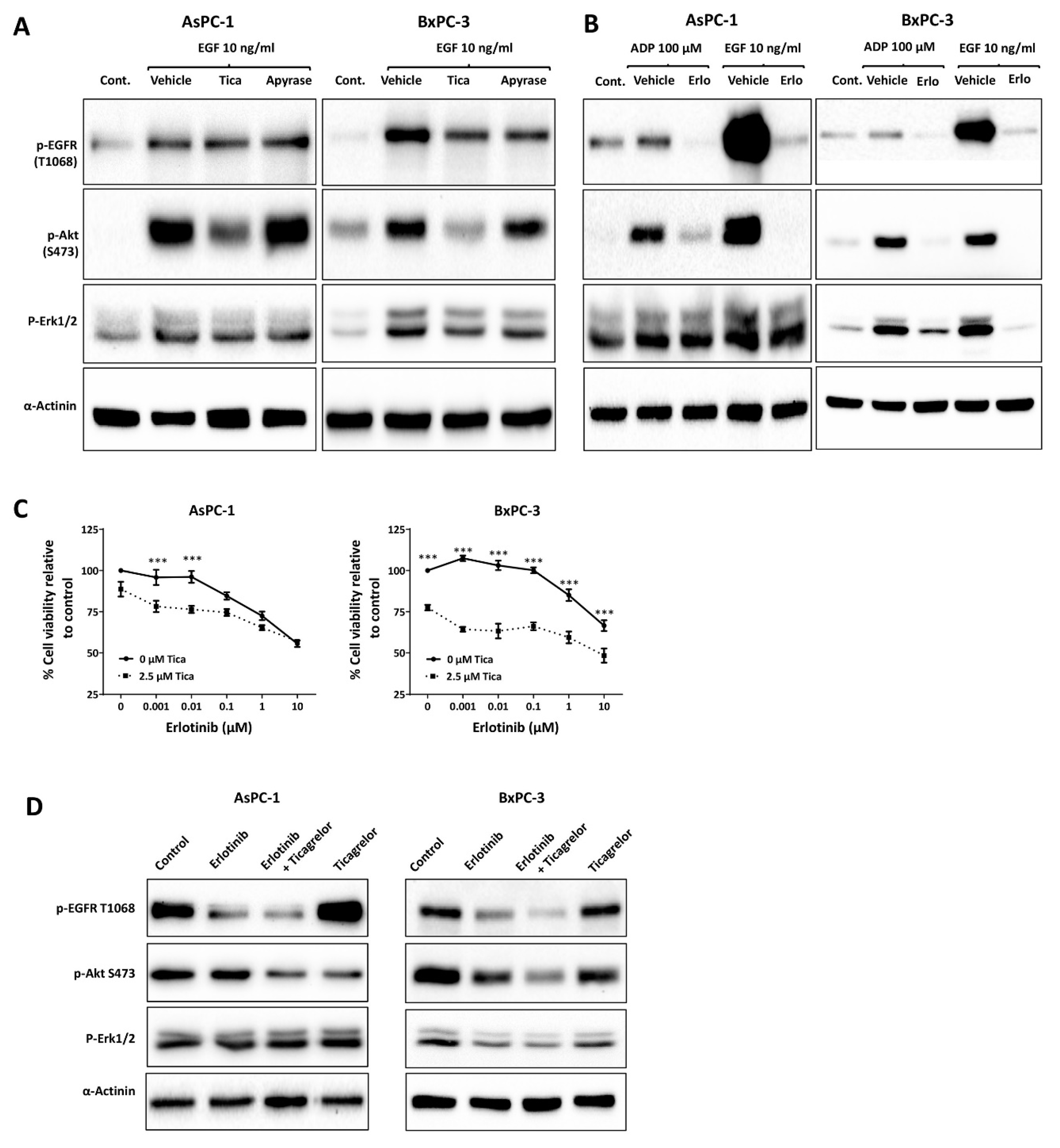
3.3. Ticagrelor Reduced EGF-Induced AKT Activation in PDAC Cells
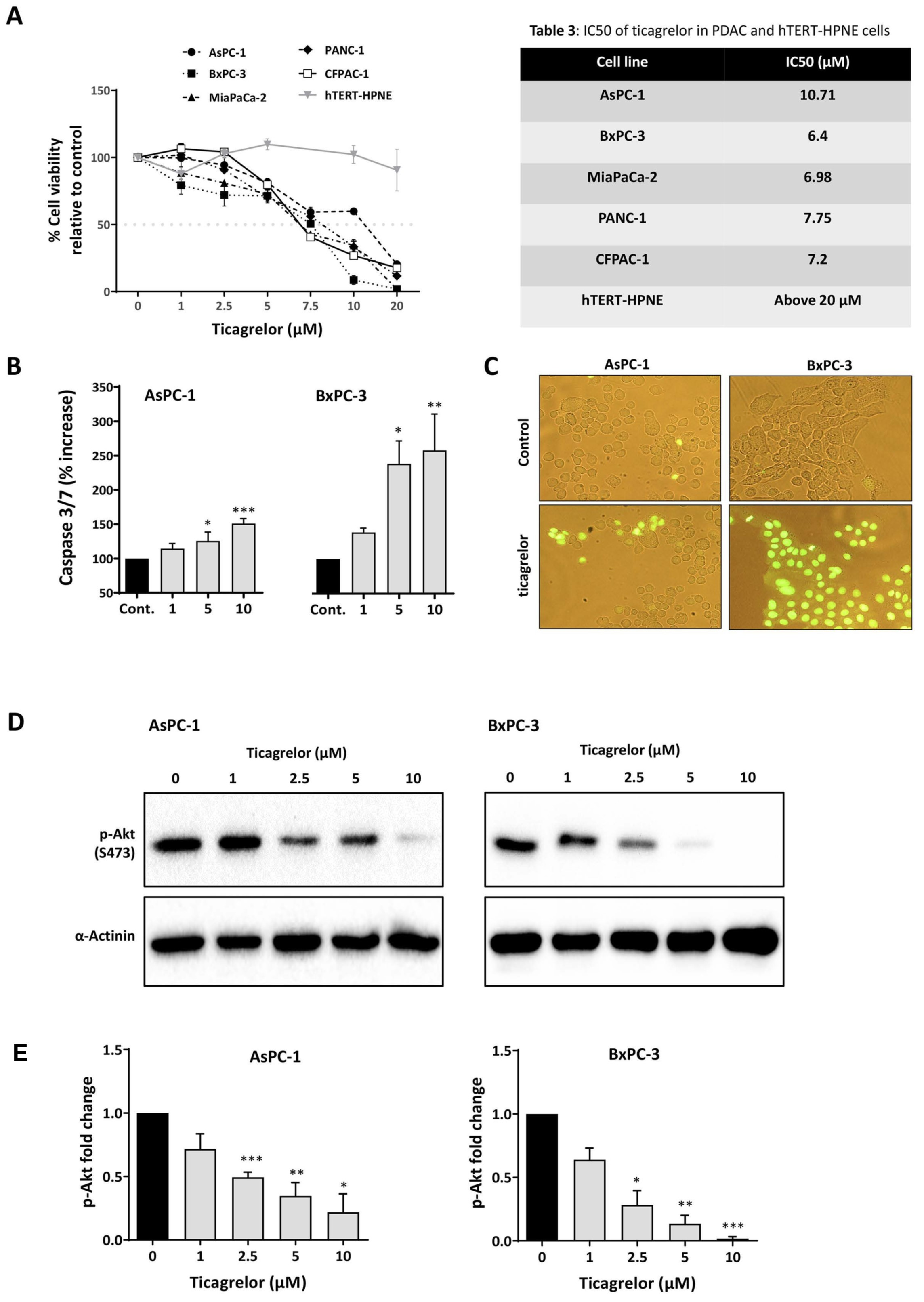
3.4. Ticagrelor Suppresses PDAC Cell Growth In Vitro
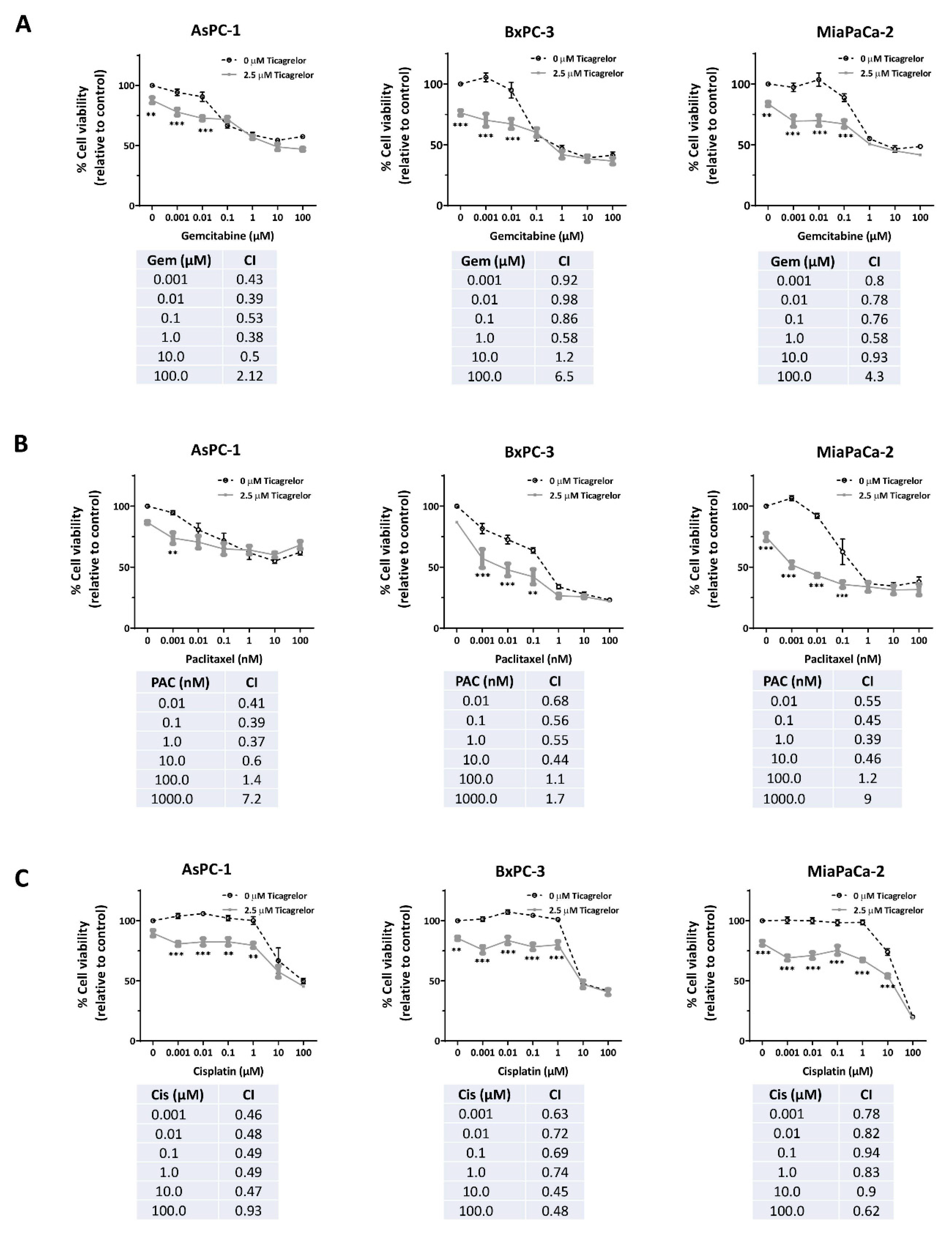
3.5. Ticagrelor Synergised with Chemotherapy in PDAC Cells In Vitro
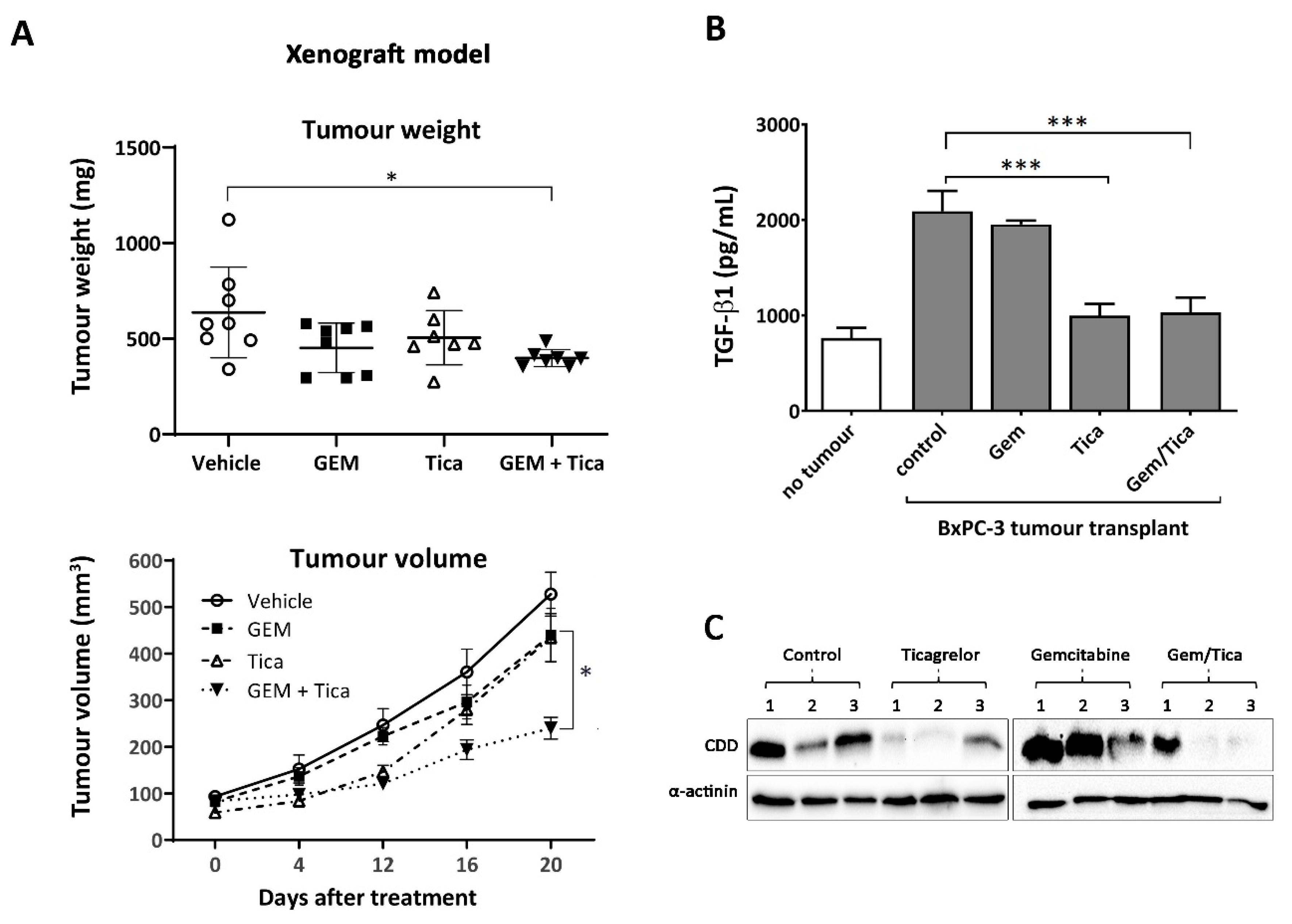
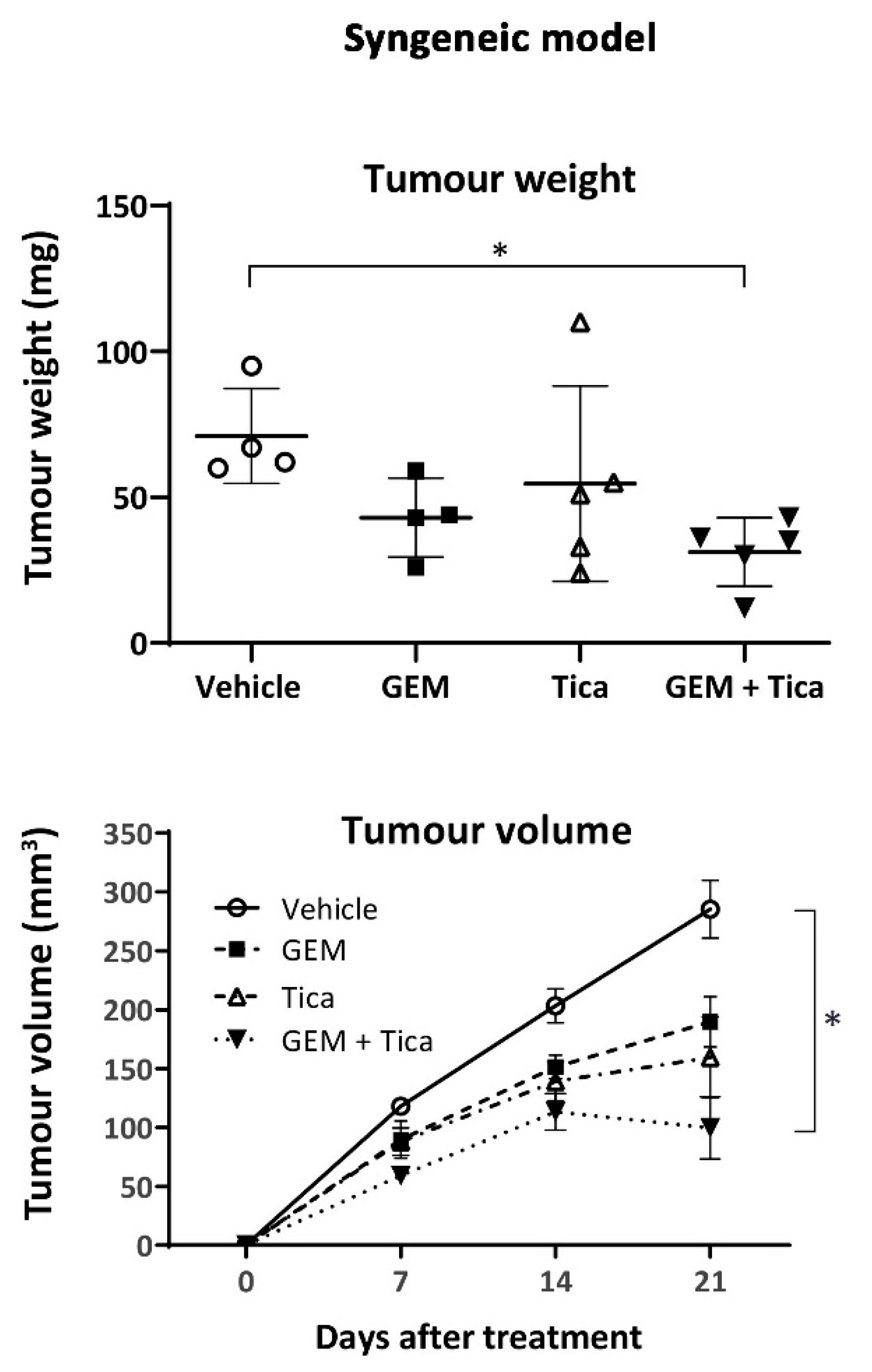
3.6. The Combination of Ticagrelor and Gemcitabine Significantly Reduced Tumour Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| CDD | Cytidine deaminase |
| CI | Combination index |
| Cis | Cisplatin |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| ENT1 | Equiliberative nucleoside transporter 1 |
| Gem | Gemcitabine |
| GEPIA | Gene expression profiling interactive analysis |
| GPCRs | G-protein coupled receptors |
| MMP | Matrix metalloproteases |
| Pac | Paclitaxel |
| PDAC | Pancreatic ductal adenocarcinoma |
| Tica | Ticagrelor |
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.P.; Sacks, G.D.; Rochefort, M.M.; Donahue, T.R.; Reber, H.A.; Tomlinson, J.S.; Dawson, D.W.; Eibl, G.; Hines, O.J. Long-term survival in patients with pancreatic ductal adenocarcinoma. Surgery 2016, 159, 1520–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khorana, A.A.; Fine, R.L. Pancreatic cancer and thromboembolic disease. Lancet Oncol. 2004, 5, 655–663. [Google Scholar] [CrossRef]
- Razak, N.A.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 2018, 10, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Abdol Razak, N.B.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87. [Google Scholar] [CrossRef]
- Agnelli, G.; Gussoni, G.; Bianchini, C.; Verso, M.; Mandalà, M.; Cavanna, L.; Barni, S.; Labianca, R.; Buzzi, F.; Scambia, G.; et al. Nadroparin for the prevention of thromboembolic events in ambulatory patients with metastatic or locally advanced solid cancer receiving chemotherapy: A randomised, placebo-controlled, double-blind study. Lancet Oncol. 2009, 10, 943–949. [Google Scholar] [CrossRef]
- Pelzer, U.; Opitz, B.; Deutschinoff, G.; Stauch, M.; Reitzig, P.C.; Hahnfeld, S.; Müller, L.; Grunewald, M.; Stieler, J.M.; Sinn, M.; et al. Efficacy of Prophylactic Low–Molecular Weight Heparin for Ambulatory Patients With Advanced Pancreatic Cancer: Outcomes From the CONKO-004 Trial. J. Clin. Oncol. 2015, 33, 2028–2034. [Google Scholar] [CrossRef]
- Klerk, C.P.; Smorenburg, S.M.; Otten, H.-M.; Lensing, A.W.; Prins, M.H.; Piovella, F.; Prandoni, P.; Bos, M.M.; Richel, D.J.; Van Tienhoven, G.; et al. The Effect of Low Molecular Weight Heparin on Survival in Patients With Advanced Malignancy. J. Clin. Oncol. 2005, 23, 2130–2135. [Google Scholar] [CrossRef]
- Elaskalani, O.; Falasca, M.; Moran, N.; Berndt, M.C.; Metharom, P. The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance. Cancers 2017, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Kunapuli, S.P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc. Natl. Acad. Sci. USA 1998, 95, 8070–8074. [Google Scholar] [CrossRef] [Green Version]
- Kahner, B.N.; Shankar, H.; Murugappan, S.; Prasad, G.L.; Kunapuli, S.P. Nucleotide receptor signaling in platelets. J. Thromb. Haemost. 2006, 4, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kunapuli, S.P. P2Y12 receptor in platelet activation. Platelets 2011, 22, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Noh, K.; Haemmerle, M.; Li, D.; Park, H.; Hu, Q.; Hisamatsu, T.; Mitamura, T.; Mak, S.L.C.; Kunapuli, S.; et al. Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 2017, 130, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Metharom, P.; Berndt, M.C.; Baker, R.I.; Andrews, R.K. Current State and Novel Approaches of Antiplatelet Therapy. Arter. Thromb. Vasc. Boil. 2015, 35, 1327–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elaskalani, O.; Berndt, M.C.; Falasca, M.; Metharom, P. Targeting Platelets for the Treatment of Cancer. Cancers 2017, 9, 94. [Google Scholar] [CrossRef]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Sham, D.; Wesley, U.V.; Hristova, M.; Van Der Vliet, A. ATP-Mediated Transactivation of the Epidermal Growth Factor Receptor in Airway Epithelial Cells Involves DUOX1-Dependent Oxidation of Src and ADAM17. PLoS ONE 2013, 8, e54391. [Google Scholar] [CrossRef] [Green Version]
- Ratchford, A.M.; Baker, O.J.; Camden, J.M.; Rikka, S.; Petris, M.J.; Seye, C.I.; Erb, L.; Weisman, G.A. P2Y2 Nucleotide Receptors Mediate Metalloprotease-dependent Phosphorylation of Epidermal Growth Factor Receptor and ErbB3 in Human Salivary Gland Cells. J. Boil. Chem. 2010, 285, 7545–7555. [Google Scholar] [CrossRef] [Green Version]
- Buvinic, S.; Bravo-Zehnder, M.; Boyer, J.L.; Huidobro-Toro, J.P.; González, A. Nucleotide P2Y1 receptor regulates EGF receptor mitogenic signaling and expression in epithelial cells. J. Cell Sci. 2007, 120, 4289–4301. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Savi, P.; Zachayus, J.-L.; Delesque-Touchard, N.; Labouret, C.; Hervé, C.; Uzabiaga, M.-F.; Pereillo, J.-M.; Culouscou, J.-M.; Bono, F.; Ferrara, P.; et al. The active metabolite of Clopidogrel disrupts P2Y12 receptor oligomers and partitions them out of lipid rafts. Proc. Natl. Acad. Sci. USA 2006, 103, 11069–11074. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Bynagari, Y.S.; Mada, S.R.; Jakubowski, J.A.; Kunapuli, S.P. Studies on the role of the extracellular cysteines and oligomeric structures of the P2Y12receptor when interacting with antagonists. J. Thromb. Haemost. 2009, 7, 232–234. [Google Scholar] [CrossRef]
- Zhang, Y.; Peti-Peterdi, J.; Müller, C.E.; Carlson, N.G.; Baqi, Y.; Strasburg, D.L.; Heiney, K.M.; Villanueva, K.; Kohan, N.E.; Kishore, B.K. P2Y12 Receptor Localizes in the Renal Collecting Duct and Its Blockade Augments Arginine Vasopressin Action and Alleviates Nephrogenic Diabetes Insipidus. J. Am. Soc. Nephrol. 2015, 26, 2978–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Li, N.; Ibrahim, S.; Smyth, E.; Woulfe, D.S. The physical association of the P2Y12 receptor with PAR4 regulates arrestin-mediated Akt activation. Mol. Pharmacol. 2014, 86, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.H.; Li, J.G.; Dores, M.R.; Trejo, J. Protease-activated receptor-4 and purinergic receptor p2y12 dimerize, co-internalize, and activate akt signaling via endosomal recruitment of beta-arrestin. J. Biol. Chem. 2017, 292, 13867–13878. [Google Scholar] [CrossRef] [Green Version]
- Rosati, A.; Basile, A.; D’Auria, R.; D’Avenia, M.; De Marco, M.; Falco, A.; Festa, M.; Guerriero, L.; Iorio, V.; Parente, R.; et al. BAG3 promotes pancreatic ductal adenocarcinoma growth by activating stromal macrophages. Nat. Commun. 2015, 6, 8695. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Dorsam, R.T.; Kunapuli, S.P. Central role of the p2y12 receptor in platelet activation. J. Clin. Investig. 2004, 113, 340–345. [Google Scholar] [CrossRef]
- Krzemiński, P.; Supłat, R.; Czajkowski, R.; Pomorski, P.; Barańska, J. Expression and functional characterization of P2Y1 and P2Y12 nucleotide receptors in long-term serum-deprived glioma C6 cells. FEBS J. 2007, 274, 1970–1982. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.P.; Maurer, B.J. Evaluating Response to Antineoplastic Drug Combinations in Tissue Culture Models. Chemosensitivity 2005, 110, 173–184. [Google Scholar]
- Food and Drug Administration (FDA). Appendix 1: Clinical pharmacology and biopharmaceutics review. In New Drug Application #22433; FDA: Silver Spring, MD, USA, 2010. [Google Scholar]
- Zhang, S.H.; Ye, J.; Zhang, Y.; Xu, X.; Liu, J.; Kunapuli, S.P.; Ding, Z. P2Y12protects platelets from apoptosis via PI3k-dependent Bak/Bax inactivation. J. Thromb. Haemost. 2013, 11, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.; Maya, J.; Butler, K. Evaluation of the pharmacokinetics and pharmacodynamics of ticagrelor co-administered with aspirin in healthy volunteers. Platelets 2013, 24, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, R.; Butler, K. A pharmacokinetic interaction study of ticagrelor and digoxin in healthy volunteers. Eur. J. Clin. Pharmacol. 2013, 69, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.B.; Jackman, L.E.; Francis, S.E.; Judge, H.M.; Nylander, S.; Storey, R.F. Ticagrelor effectively and reversibly blocks murine platelet P2Y12-mediated thrombosis and demonstrates a requirement for sustained P2Y12 inhibition to prevent subsequent neointima. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2385–2391. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Corrie, P.; Qian, W.; Gopinathan, A.; Williams, M.; Brais, R.; Valle, J.W.; Basu, B.; Falk, S.; Iwuji, C.; Wasan, H.; et al. Strong tumour cytidine deaminase (CDA) staining predicts for improved survival associated with sequential nab-paclitaxel (NABP) and gemcitabine (GEM) chemotherapy as first line treatment of patients (PTS) with metastatic pancreatic adenocarcinoma (MPDAC). Ann. Oncol. 2017, 28, 73P. [Google Scholar] [CrossRef] [Green Version]
- Elaskalani, O.; Metharom, P.; (Curtin University, Perth, Australia). Personal Communication, 2019.
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta Pharm. Sin. B 2018, 8, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Pierce, K.L.; Zamah, A.M.; Miller, W.E.; Ahn, S.; Daaka, Y.; Lefkowitz, R.J.; Luttrell, L.M. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J. Boil. Chem. 2000, 275, 9572–9580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ramirez, A.S.; Diaz-Munoz, M.; Butanda-Ochoa, A.; Vazquez-Cuevas, F.G. Nucleotides and nucleoside signaling in the regulation of the epithelium to mesenchymal transition (emt). Purinergic Signal. 2017, 13, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-H.; Qiu, Y.; Zhang, H.-Q.; Liu, Y.; You, J.-F.; Tian, X.-X.; Fang, W.-G. P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br. J. Cancer 2013, 109, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jin, J.; Kunapuli, S.P. Akt activation in platelets depends on gi signaling pathways. J. Biol. Chem. 2004, 279, 4186–4195. [Google Scholar] [CrossRef] [Green Version]
- Aungraheeta, R.; Conibear, A.; Butler, M.; Kelly, E.; Nylander, S.; Mumford, A.; Mundell, S.J. Inverse agonism at the P2Y12 receptor and ENT1 transporter blockade contribute to platelet inhibition by ticagrelor. Blood 2016, 128, 2717–2728. [Google Scholar] [CrossRef]
- Xie, K.-Q.; Zhang, L.-M.; Cao, Y.; Zhu, J.; Feng, L.-Y. Adenosine A1 receptor-mediated transactivation of the EGF receptor produces a neuroprotective effect on cortical neurons in vitro. Acta Pharmacol. Sin. 2009, 30, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Reiner, M.F.; Breitenstein, A.; Holy, E.W.; Glanzmann, M.; Amstalden, H.; Stämpfli, S.F.; Bonetti, N.R.; Falk, V.; Keller, S.; Savarese, G.; et al. Ticagrelor, but not clopidogrel active metabolite, displays antithrombotic properties in the left atrial endocardium. Eur. Hear. J. 2017, 38, 916–919. [Google Scholar] [CrossRef]
- Cheng, A.N.; Lo, Y.-K.; Lin, Y.-S.; Tang, T.-K.; Hsu, C.-H.; Hsu, J.T.-A.; Lee, A.Y.-L. Identification of Novel Cdc7 Kinase Inhibitors as Anti-Cancer Agents that Target the Interaction with Dbf4 by the Fragment Complementation and Drug Repositioning Approach. EBioMedicine 2018, 36, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert t cell immunity against cancer via garp-tgfbeta axis. Sci. Immunol. 2017, 2, 7911. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. AsPC-1 | B. BxPC-3 | ||
|---|---|---|---|
| Erlotinib (µM) | CI | Erlotinib (µM) | CI |
| 0.001 | 0.43 | 0.001 | 0.88 |
| 0.01 | 0.42 | 0.01 | 0.84 |
| 0.1 | 0.5 | 0.1 | 0.91 |
| 1 | 0.62 | 1 | 0.8 |
| 10 | 1.4 | 10 | 1.1 |
| Cell Line | IC50 (µM) |
|---|---|
| AsPC-1 | 10.71 |
| BxPC-3 | 6.4 |
| MiaPaCa-2 | 6.98 |
| PANC-1 | 7.75 |
| CFPAC-1 | 7.2 |
| hTERT-HPNE | Above 20 µM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elaskalani, O.; Domenichini, A.; Abdol Razak, N.B.; E. Dye, D.; Falasca, M.; Metharom, P. Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells. Cancers 2020, 12, 250. https://doi.org/10.3390/cancers12010250
Elaskalani O, Domenichini A, Abdol Razak NB, E. Dye D, Falasca M, Metharom P. Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells. Cancers. 2020; 12(1):250. https://doi.org/10.3390/cancers12010250
Chicago/Turabian StyleElaskalani, Omar, Alice Domenichini, Norbaini Binti Abdol Razak, Danielle E. Dye, Marco Falasca, and Pat Metharom. 2020. "Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells" Cancers 12, no. 1: 250. https://doi.org/10.3390/cancers12010250
APA StyleElaskalani, O., Domenichini, A., Abdol Razak, N. B., E. Dye, D., Falasca, M., & Metharom, P. (2020). Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells. Cancers, 12(1), 250. https://doi.org/10.3390/cancers12010250