Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope



Abstract
:1. Introduction
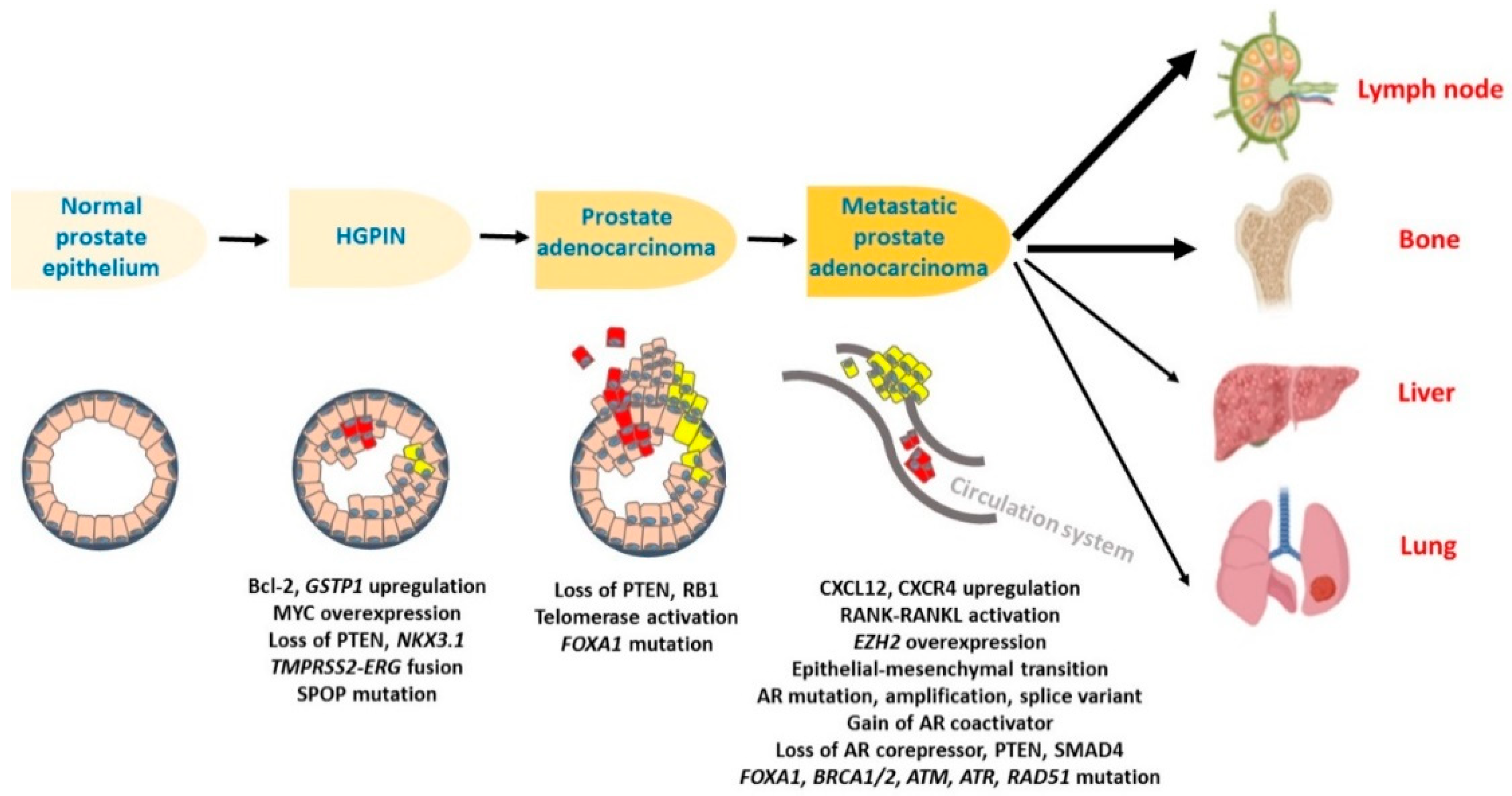
2. The Cellular and Molecular Progression of Prostate Cancer
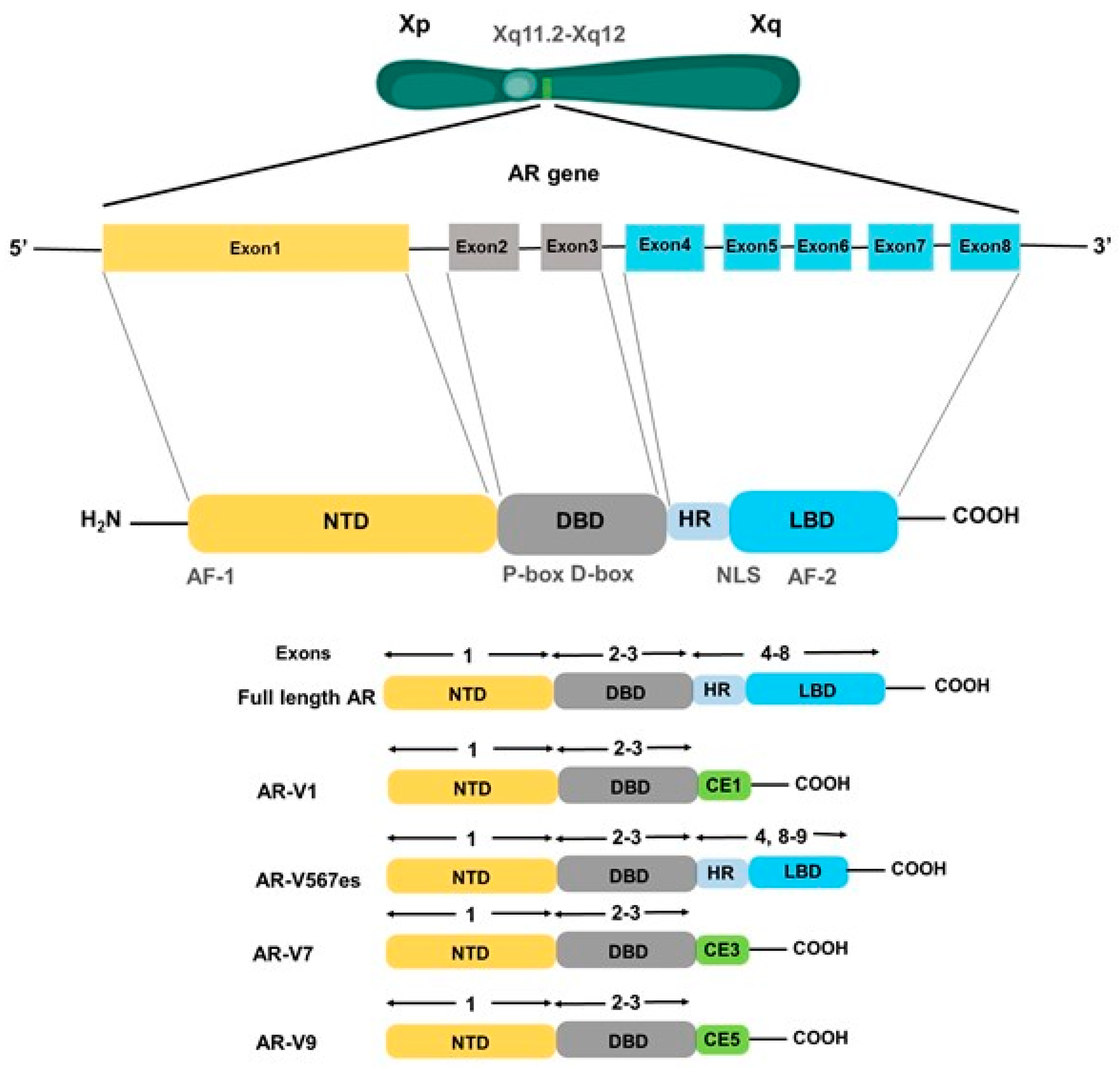
3. Androgen Receptor: Structure and Mechanisms of Action
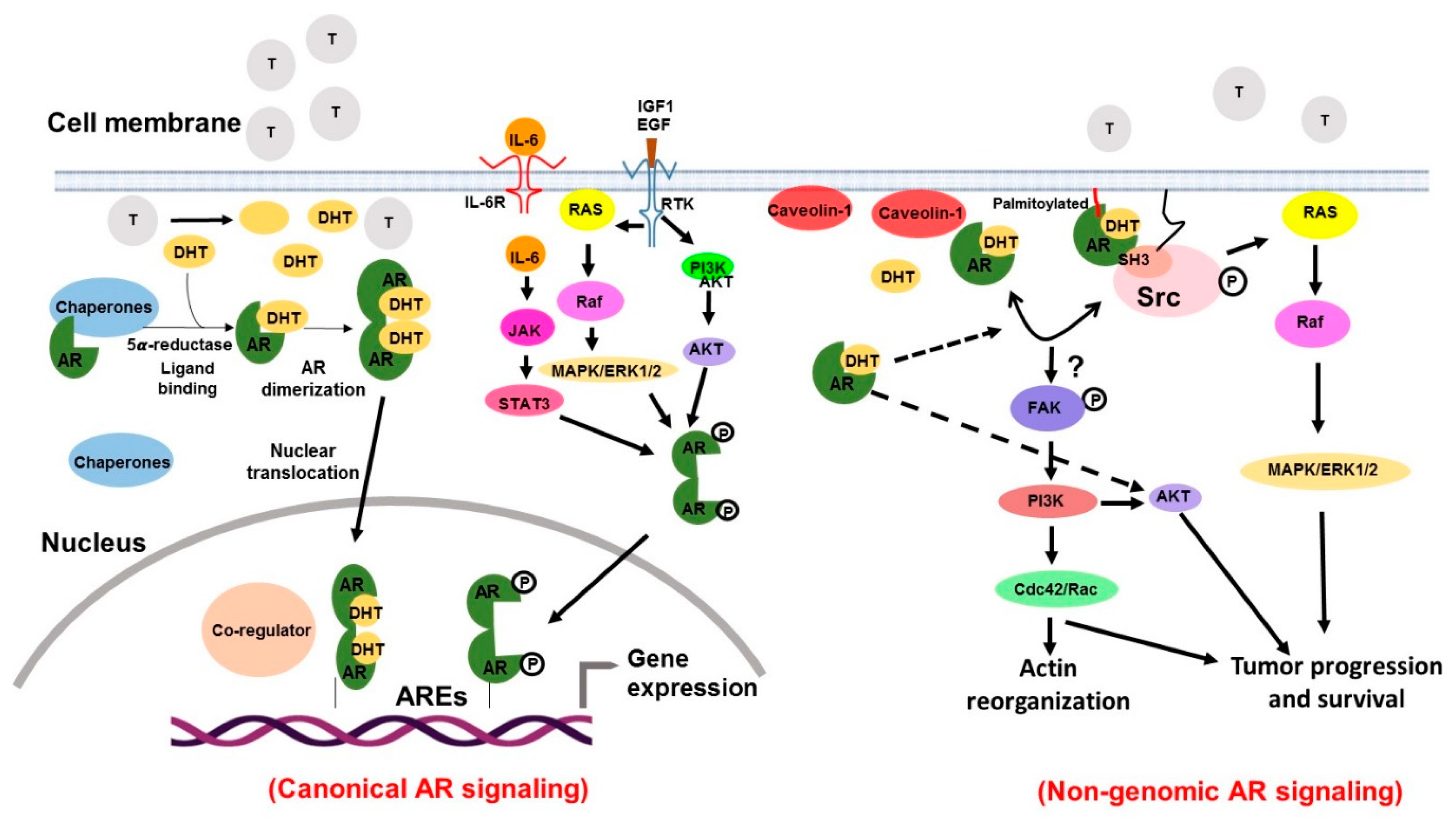
4. Evidence Implicating Androgen Receptor Signaling in Prostate Cancer Pathogenesis
4.1. AR Mutations
4.2. AR Amplification/Overexpression and Alternative Splicing
4.3. Altered Expression of AR Co-Regulators
4.4. Androgen Biosynthesis
5. Established and Evolving Therapies Targeting Androgen Receptor Signaling in Prostate Cancer
5.1. Targeting the Ligand
5.1.1. LHRH Agonists
5.1.2. LHRH Antagonists
5.1.3. Androgen Synthesis Inhibitor
5.2. Targeting the Receptor
5.2.1. Steroidal Anti-Androgens
5.2.2. Non-Steroidal Anti-Androgens
5.3. Targeting the AR Interaction with Co-Regulators
5.4. Targeting by AR Signaling by the Natural Agents
6. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Benjamins, M.R.; Hunt, B.R.; Raleigh, S.M.; Hirschtick, J.L.; Hughes, M.M. Racial Disparities in Prostate Cancer Mortality in the 50 Largest US Cities. Cancer Epidemiol. 2016, 44, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, D.; Packianathan, S.; Chen, A.M.; Vijayakumar, S. Do African-American men need separate prostate cancer screening guidelines? BMC Urol. 2016, 16, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Vignera, S.; Condorelli, R.A.; Russo, G.I.; Morgia, G.; Calogero, A.E. Endocrine control of benign prostatic hyperplasia. Andrology 2016, 4, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V. Targeting androgen receptor action for prostate cancer treatment: does the post-receptor level provide novel opportunities? Int. J. Biol. Sci. 2014, 10, 576–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Heiss, C.E. Inhibition of 5 alpha-reductase, receptor binding, and nuclear uptake of androgens in the prostate by a 4-methyl-4-aza-steroid. J. Biol. Chem. 1981, 256, 7998–8005. [Google Scholar] [PubMed]
- Phillips, R. Prostate cancer: Novel targeting of androgen signalling in CRPC. Nat. Rev. Urol. 2014, 11, 303. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Timms, B.G. Prostate development: A historical perspective. Differentiation 2008, 76, 565–577. [Google Scholar] [CrossRef]
- Haffner, J.; Potiron, E.; Bouye, S.; Puech, P.; Leroy, X.; Lemaitre, L.; Villers, A. Peripheral zone prostate cancers: location and intraprostatic patterns of spread at histopathology. Prostate 2009, 69, 276–282. [Google Scholar] [CrossRef] [PubMed]
- van Leenders, G.J.; Schalken, J.A. Epithelial cell differentiation in the human prostate epithelium: implications for the pathogenesis and therapy of prostate cancer. Crit. Rev. Oncol. Hematol. 2003, 46, S3–S10. [Google Scholar] [CrossRef]
- Garber, K. A tale of two cells: Discovering the origin of prostate cancer. J. Natl. Cancer Inst. 2010, 102, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, T.; Cooper, A.R.; Drake, J.M.; Liu, X.; Armstrong, A.J.; Pienta, K.J.; Zhang, H.; Kohn, D.B.; Huang, J.; Witte, O.N.; et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20111–20116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal cells are favored as the cell of origin for prostate cancer. Cell Rep. 2014, 8, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostwick, D.G.; Cheng, L. Precursors of prostate cancer. Histopathology 2012, 60, 4–27. [Google Scholar] [CrossRef]
- Ayala, A.G.; Ro, J.Y. Prostatic intraepithelial neoplasia: recent advances. Arch. Pathol. Lab. Med. 2007, 131, 1257–1266. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Liu, L.; Brawer, M.K.; Qian, J. High-grade prostatic intraepithelial neoplasia. Rev. Urol. 2004, 6, 171–179. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Marchi, V.L.; Epstein, J.I.; Nelson, W.G. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am. J. Pathol. 1999, 155, 1985–1992. [Google Scholar] [CrossRef]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta 2016, 1863, 1238–1260. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Bjartell, A.; Chinnaiyan, A.M.; Jenster, G.; Nam, R.K.; Rubin, M.A.; Schalken, J.A. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur. Urol. 2009, 56, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Xin, L.; Goldstein, A.S.; Lawson, D.A.; Teitell, M.A.; Witte, O.N. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12465–12470. [Google Scholar] [CrossRef] [Green Version]
- Bolton, E.M.; Tuzova, A.V.; Walsh, A.L.; Lynch, T.; Perry, A.S. Noncoding RNAs in prostate cancer: The long and the short of it. Clin. Cancer Res. 2014, 20, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Faber, P.W.; van Rooij, H.C.; van der Korput, J.A.; Ris-Stalpers, C.; Klaassen, P.; Trapman, J.; Brinkmann, A.O. Structural organization of the human androgen receptor gene. J. Mol. Endocrinol. 1989, 2, R1–R4. [Google Scholar] [CrossRef]
- Lubahn, D.B.; Brown, T.R.; Simental, J.A.; Higgs, H.N.; Migeon, C.J.; Wilson, E.M.; French, F.S. Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proc. Natl. Acad. Sci. USA 1989, 86, 9534–9538. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E.; Stampfer, M.J.; Krithivas, K.; Brown, M.; Dahl, D.; Brufsky, A.; Talcott, J.; Hennekens, C.H.; Kantoff, P.W. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 3320–3323. [Google Scholar] [CrossRef] [Green Version]
- Schoenmakers, E.; Alen, P.; Verrijdt, G.; Peeters, B.; Verhoeven, G.; Rombauts, W.; Claessens, F. Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem. J. 1999, 341, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, J.W.; Chapman, L.; Finch, J.T.; Rhodes, D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 1993, 75, 567–578. [Google Scholar] [CrossRef]
- Alen, P.; Claessens, F.; Verhoeven, G.; Rombauts, W.; Peeters, B. The androgen receptor amino-terminal domain plays a key role in p160 coactivator-stimulated gene transcription. Mol. Cell. Biol. 1999, 19, 6085–6097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H.; et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol. Cancer Res. 2017, 15, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen Receptor Variant AR-V9 Is Coexpressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Sprenger, C.; Sun, S.; Epilepsia, K.S.; Haugk, K.; Zhang, X.; Coleman, I.; Nelson, P.S.; Plymate, S. AR variant ARv567es induces carcinogenesis in a novel transgenic mouse model of prostate cancer. Neoplasia 2013, 15, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, M.; Obara, T.; Sasano, S.; Onuki, T. Is taking preoperative high-dose steroid necessary? Eur. J. Cardiothorac. Surg. 2006, 30, 688–689. [Google Scholar] [CrossRef]
- Niu, Y.; Chang, T.M.; Yeh, S.; Ma, W.L.; Wang, Y.Z.; Chang, C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene 2010, 29, 3593–3604. [Google Scholar] [CrossRef] [Green Version]
- Heery, D.M.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef]
- Ueda, T.; Bruchovsky, N.; Sadar, M.D. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J. Biol. Chem. 2002, 277, 7076–7085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traish, A.M.; Morgentaler, A. Epidermal growth factor receptor expression escapes androgen regulation in prostate cancer: a potential molecular switch for tumour growth. Br. J. Cancer 2009, 101, 1949–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; Auricchio, F. Analysis of androgen receptor rapid actions in cellular signaling pathways: receptor/Src association. Methods Mol. Biol. 2011, 776, 361–370. [Google Scholar] [CrossRef]
- Lu, M.L.; Schneider, M.C.; Zheng, Y.; Zhang, X.; Richie, J.P. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J. Biol. Chem. 2001, 276, 13442–13451. [Google Scholar] [CrossRef] [Green Version]
- Pedram, A.; Razandi, M.; Sainson, R.C.; Kim, J.K.; Hughes, C.C.; Levin, E.R. A conserved mechanism for steroid receptor translocation to the plasma membrane. J. Biol. Chem. 2007, 282, 22278–22288. [Google Scholar] [CrossRef] [Green Version]
- Kalyvianaki, K.; Panagiotopoulos, A.A.; Malamos, P.; Moustou, E.; Tzardi, M.; Stathopoulos, E.N.; Ioannidis, G.S.; Marias, K.; Notas, G.; Theodoropoulos, P.A.; et al. Membrane androgen receptors (OXER1, GPRC6A AND ZIP9) in prostate and breast cancer: A comparative study of their expression. Steroids 2019, 142, 100–108. [Google Scholar] [CrossRef]
- Papakonstanti, E.A.; Kampa, M.; Castanas, E.; Stournaras, C. A rapid, nongenomic, signaling pathway regulates the actin reorganization induced by activation of membrane testosterone receptors. Mol. Endocrinol. 2003, 17, 870–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Sato, N.; Watabe, Y.; Masai, M.; Seino, S.; Shimazaki, J. Androgen receptor gene mutations in human prostate cancer. J. Steroid Biochem. Mol. Biol. 1993, 46, 759–765. [Google Scholar] [CrossRef]
- van de Wijngaart, D.J.; Molier, M.; Lusher, S.J.; Hersmus, R.; Jenster, G.; Trapman, J.; Dubbink, H.J. Systematic structure-function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J. Biol. Chem. 2010, 285, 5097–5105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steketee, K.; Timmerman, L.; Ziel-van der Made, A.C.; Doesburg, P.; Brinkmann, A.O.; Trapman, J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int. J. Cancer 2002, 100, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Wilding, G.; Chen, M.; Gelmann, E.P. Aberrant response in vitro of hormone-responsive prostate cancer cells to antiandrogens. Prostate 1989, 14, 103–115. [Google Scholar] [CrossRef]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [Green Version]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef]
- Kawata, H.; Ishikura, N.; Watanabe, M.; Nishimoto, A.; Tsunenari, T.; Aoki, Y. Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate 2010, 70, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhani, N.; Generali, D.; D’Angelo, A.; Aieta, M.; Roviello, G. Current status of androgen receptor-splice variant 7 inhibitor niclosamide in castrate-resistant prostate-cancer. Investig. New Drugs 2018, 36, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Onstenk, W.; Sieuwerts, A.M.; Kraan, J.; Van, M.; Nieuweboer, A.J.; Mathijssen, R.H.; Hamberg, P.; Meulenbeld, H.J.; De Laere, B.; Dirix, L.Y.; et al. Efficacy of Cabazitaxel in Castration-resistant Prostate Cancer Is Independent of the Presence of AR-V7 in Circulating Tumor Cells. Eur. Urol. 2015, 68, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.; Dong, Y.; Zhang, H.; Qi, Y.; Balk, S.P.; Sartor, O. Castration-resistant prostate cancer: Adaptive responses in the androgen axis. Cancer Treat. Rev. 2014, 40, 426–433. [Google Scholar] [CrossRef]
- Agoulnik, I.U.; Vaid, A.; Bingman, W.E., 3rd; Erdeme, H.; Frolov, A.; Smith, C.L.; Ayala, G.; Ittmann, M.M.; Weigel, N.L. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005, 65, 7959–7967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.J.; Yan, J.; Luo, W.; Ayala, G.; Lin, S.H.; Erdem, H.; Ittmann, M.; Tsai, S.Y.; Tsai, M.J. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005, 65, 7976–7983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, J.C.; Liu, Z.; Liao, L.; Wang, F.; Xu, Y.; Wu, Y.L.; Zhou, N.; Ittmann, M.; Xu, J. The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer. Cancer Res. 2013, 73, 3997–4008. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Hashimoto, Y.; Kwak, I.; Tsai, S.Y.; Tsai, M.J. Role of the steroid receptor coactivator SRC-3 in cell growth. Mol. Cell. Biol. 2003, 23, 7742–7755. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.C.; Yeh, S.; Yeh, S.D.; Sampson, E.R.; Huang, J.; Li, P.; Hsu, C.L.; Ting, H.J.; Lin, H.K.; Wang, L.; et al. Functional domain and motif analyses of androgen receptor coregulator ARA70 and its differential expression in prostate cancer. J. Biol. Chem. 2004, 279, 33438–33446. [Google Scholar] [CrossRef] [Green Version]
- Yeh, S.; Chang, C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc. Natl. Acad. Sci. USA 1996, 93, 5517–5521. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.X.; He, B.; Hall, S.H.; Wilson, E.M.; French, F.S. Domain interactions between coregulator ARA(70) and the androgen receptor (AR). Mol. Endocrinol. 2002, 16, 287–300. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Penning, T.M. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab. 2010, 21, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M. Androgen biosynthesis in castration-resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, T67–T78. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, K.E.; Kelly, W.K. Outsmarting androgen receptor: creative approaches for targeting aberrant androgen signaling in advanced prostate cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 483–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, R.S.; Fitzpatrick, J.M.; Clarke, N. Abarelix and other gonadotrophin-releasing hormone antagonists in prostate cancer. BJU Int. 2009, 104, 1580–1584. [Google Scholar] [CrossRef]
- Roila, F. Buserelin in the treatment of prostatic cancer. Biomed. Pharmacother. 1989, 43, 279–285. [Google Scholar] [CrossRef]
- Wilson, A.C.; Meethal, S.V.; Bowen, R.L.; Atwood, C.S. Leuprolide acetate: A drug of diverse clinical applications. Expert Opin. Investig. Drugs 2007, 16, 1851–1863. [Google Scholar] [CrossRef]
- Chrisp, P.; Goa, K.L. Goserelin. A review of its pharmacodynamic and pharmacokinetic properties, and clinical use in sex hormone-related conditions. Drugs 1991, 41, 254–288. [Google Scholar] [CrossRef]
- Brogden, R.N.; Faulds, D. Goserelin. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in prostate cancer. Drugs Aging 1995, 6, 324–343. [Google Scholar] [CrossRef]
- Djavan, B.; Schlegel, P.; Salomon, G.; Eckersberger, E.; Sadri, H.; Graefen, M. Analysis of testosterone suppression in men receiving histrelin, a novel GnRH agonist for the treatment of prostate cancer. Can. J. Urol. 2010, 17, 5265–5271. [Google Scholar]
- Deeks, E.D. Histrelin: in advanced prostate cancer. Drugs 2010, 70, 623–630. [Google Scholar] [CrossRef]
- Lahlou, N.; Carel, J.C.; Chaussain, J.L.; Roger, M. Pharmacokinetics and pharmacodynamics of GnRH agonists: clinical implications in pediatrics. J. Pediatr. Endocrinol. Metab. 2000, 13, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Ploussard, G.; Mongiat-Artus, P. Triptorelin in the management of prostate cancer. Future Oncol. 2013, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Debruyne, F.M. Gonadotropin-releasing hormone antagonist in the management of prostate cancer. Rev. Urol. 2004, 6, S25–S32. [Google Scholar] [PubMed]
- Castellon, E.; Clementi, M.; Hitschfeld, C.; Sanchez, C.; Benitez, D.; Saenz, L.; Contreras, H.; Huidobro, C. Effect of leuprolide and cetrorelix on cell growth, apoptosis, and GnRH receptor expression in primary cell cultures from human prostate carcinoma. Cancer Investig. 2006, 24, 261–268. [Google Scholar] [CrossRef]
- Oberye, J.; Mannaerts, B.; Huisman, J.; Timmer, C. Local tolerance, pharmacokinetics, and dynamics of ganirelix (Orgalutran) administration by Medi-Jector compared to conventional needle injections. Hum. Reprod. 2000, 15, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, M. Degarelix: A gonadotropin-releasing hormone antagonist for the management of prostate cancer. Clin. Ther. 2009, 31, 2312–2331. [Google Scholar] [CrossRef]
- O’Donnell, A.; Judson, I.; Dowsett, M.; Raynaud, F.; Dearnaley, D.; Mason, M.; Harland, S.; Robbins, A.; Halbert, G.; Nutley, B.; et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br. J. Cancer 2004, 90, 2317–2325. [Google Scholar] [CrossRef]
- Yamaoka, M.; Hara, T.; Hitaka, T.; Kaku, T.; Takeuchi, T.; Takahashi, J.; Asahi, S.; Miki, H.; Tasaka, A.; Kusaka, M. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 2012, 129, 115–128. [Google Scholar] [CrossRef]
- Vaughan, E.D. Long-Term Experience with 5-alpha-Reductase Inhibitors. Rev. Urol. 2003, 5, S28–S33. [Google Scholar]
- Goldenberg, S.L.; Bruchovsky, N. Use of cyproterone acetate in prostate cancer. Urol. Clin. North Am. 1991, 18, 111–122. [Google Scholar]
- Orme, L.M.; Bond, J.D.; Humphrey, M.S.; Zacharin, M.R.; Downie, P.A.; Jamsen, K.M.; Mitchell, S.L.; Robinson, J.M.; Grapsas, N.A.; Ashley, D.M. Megestrol acetate in pediatric oncology patients may lead to severe, symptomatic adrenal suppression. Cancer 2003, 98, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Seeger, H.; Mueck, A.O. The pharmacology of dienogest. Maturitas 2012, 71, 337–344. [Google Scholar] [CrossRef] [PubMed]
- McClurg, U.L.; Azizyan, M.; Dransfield, D.T.; Namdev, N.; Chit, N.; Nakjang, S.; Robson, C.N. The novel anti-androgen candidate galeterone targets deubiquitinating enzymes, USP12 and USP46, to control prostate cancer growth and survival. Oncotarget 2018, 9, 24992–25007. [Google Scholar] [CrossRef]
- Terouanne, B.; Paris, F.; Servant, N.; Georget, V.; Sultan, C. Evidence that chlormadinone acetate exhibits antiandrogenic activity in androgen-dependent cell line. Mol. Cell. Endocrinol. 2002, 198, 143–147. [Google Scholar] [CrossRef]
- Laufer, M.; Sinibaldi, V.J.; Carducci, M.A.; Eisenberger, M.A. Rapid disease progression after the administration of bicalutamide in patients with metastatic prostate cancer. Urology 1999, 54, 745. [Google Scholar] [CrossRef]
- Shet, M.S.; McPhaul, M.; Fisher, C.W.; Stallings, N.R.; Estabrook, R.W. Metabolism of the antiandrogenic drug (Flutamide) by human CYP1A2. Drug Metab. Dispos. 1997, 25, 1298–1303. [Google Scholar]
- Schasfoort, E.M.; Van De Beek, C.; Newling, D.W. Safety and efficacy of a non-steroidal anti-androgen, based on results of a post marketing surveillance of nilutamide. Prostate Cancer Prostatic Dis. 2001, 4, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, J.A.; de Vries, M.; Krauwinkel, W.; Ohtsu, Y.; Noukens, J.; van der Walt, J.S.; Mol, R.; Mordenti, J.; Ouatas, T. Pharmacokinetic Drug Interaction Studies with Enzalutamide. Clin. Pharmacokinet. 2015, 54, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Al-Salama, Z.T. Apalutamide: First Global Approval. Drugs 2018, 78, 699–705. [Google Scholar] [CrossRef]
- Loddick, S.A.; Ross, S.J.; Thomason, A.G.; Robinson, D.M.; Walker, G.E.; Dunkley, T.P.; Brave, S.R.; Broadbent, N.; Stratton, N.C.; Trueman, D.; et al. AZD3514: A small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 1715–1727. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Albiges, L.; Loriot, Y.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, Y.; Chow, D.C.; Yan, F.; Hsu, C.C.; Stossi, F.; Mancini, M.A.; Palzkill, T.; Liao, L.; Zhou, S.; et al. Characterization of a Steroid Receptor Coactivator Small Molecule Stimulator that Overstimulates Cancer Cells and Leads to Cell Stress and Death. Cancer Cell 2015, 28, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, N.; Di Lorenzo, G.; Sonpavde, G.; Bellmunt, J. New agents for prostate cancer. Ann. Oncol. 2014, 25, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, K.J.; Langmann, G.; Ai, J.; Ramos-Garcia, R.; Vessella, R.L.; Wang, Z. Hsp90 inhibitor 17-AAG inhibits progression of LuCaP35 xenograft prostate tumors to castration resistance. Prostate 2012, 72, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Kumar, V.; Lee, S.; Iwai, A.; Neckers, L.; Malhotra, S.V.; Trepel, J.B. Methoxychalcone inhibitors of androgen receptor translocation and function. Bioorg. Med. Chem. Lett. 2012, 22, 2105–2109. [Google Scholar] [CrossRef] [Green Version]
- Ferraldeschi, R.; Welti, J.; Powers, M.V.; Yuan, W.; Smyth, T.; Seed, G.; Riisnaes, R.; Hedayat, S.; Wang, H.; Crespo, M.; et al. Second-Generation HSP90 Inhibitor Onalespib Blocks mRNA Splicing of Androgen Receptor Variant 7 in Prostate Cancer Cells. Cancer Res. 2016, 76, 2731–2742. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Shida, Y.; Hakariya, T.; Sakai, H. Anti-Cancer Effects of Green Tea Polyphenols Against Prostate Cancer. Molecules 2019, 24, 193. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Slivova, V.; Valachovicova, T.; Harvey, K.; Sliva, D. Ganoderma lucidum inhibits proliferation and induces apoptosis in human prostate cancer cells PC-3. Int. J. Oncol. 2004, 24, 1093–1099. [Google Scholar] [CrossRef]
- Shetty, A.V.; Thirugnanam, S.; Dakshinamoorthy, G.; Samykutty, A.; Zheng, G.; Chen, A.; Bosland, M.C.; Kajdacsy-Balla, A.; Gnanasekar, M. 18alpha-glycyrrhetinic acid targets prostate cancer cells by down-regulating inflammation-related genes. Int. J. Oncol. 2011, 39, 635–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, K.T.; Figg, W.D. The potential role of curcumin in prostate cancer: the importance of optimizing pharmacokinetics in clinical studies. Transl. Cancer Res. 2016, 5, S1107–S1110. [Google Scholar] [CrossRef]
- Meimetis, L.G.; Williams, D.E.; Mawji, N.R.; Banuelos, C.A.; Lal, A.A.; Park, J.J.; Tien, A.H.; Fernandez, J.G.; de Voogd, N.J.; Sadar, M.D.; et al. Niphatenones, glycerol ethers from the sponge Niphates digitalis block androgen receptor transcriptional activity in prostate cancer cells: structure elucidation, synthesis, and biological activity. J. Med. Chem. 2012, 55, 503–514. [Google Scholar] [CrossRef]
- Banuelos, C.A.; Tavakoli, I.; Tien, A.H.; Caley, D.P.; Mawji, N.R.; Li, Z.; Wang, J.; Yang, Y.C.; Imamura, Y.; Yan, L.; et al. Sintokamide A Is a Novel Antagonist of Androgen Receptor That Uniquely Binds Activation Function-1 in Its Amino-terminal Domain. J. Biol. Chem. 2016, 291, 22231–22243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayehmiri, K.; Azami, M.; Mohammadi, Y.; Soleymani, A.; Tardeh, Z. The association between Selenium and Prostate Cancer: a Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2018, 19, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Mir, H.; Kapur, N.; Gales, D.N.; Carriere, P.P.; Singh, S. Quercetin inhibits prostate cancer by attenuating cell survival and inhibiting anti-apoptotic pathways. World J. Surg. Oncol. 2018, 16, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Du, S.; Wang, J. Berberine inhibits the proliferation of prostate cancer cells and induces G(0)/G(1) or G(2)/M phase arrest at different concentrations. Mol. Med. Rep. 2015, 11, 3920–3924. [Google Scholar] [CrossRef] [Green Version]
- Zubair, H.; Bhardwaj, A.; Ahmad, A.; Srivastava, S.K.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Hydroxytyrosol Induces Apoptosis and Cell Cycle Arrest and Suppresses Multiple Oncogenic Signaling Pathways in Prostate Cancer Cells. Nutr. Cancer 2017, 69, 932–942. [Google Scholar] [CrossRef]
- Crawford, E.D. Hormonal therapy in prostate cancer: historical approaches. Rev. Urol. 2004, 6, S3–S11. [Google Scholar]
- Xu, Y.; Jiang, Y.F.; Wu, B. New agonist- and antagonist-based treatment approaches for advanced prostate cancer. J. Int. Med. Res. 2012, 40, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Labrie, F.; Dupont, A.; Belanger, A.; Lachance, R. Flutamide eliminates the risk of disease flare in prostatic cancer patients treated with a luteinizing hormone-releasing hormone agonist. J. Urol. 1987, 138, 804–806. [Google Scholar] [CrossRef]
- Rhee, H.; Gunter, J.H.; Heathcote, P.; Ho, K.; Stricker, P.; Corcoran, N.M.; Nelson, C.C. Adverse effects of androgen-deprivation therapy in prostate cancer and their management. BJU Int. 2015, 115, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Thompson, I.M. Flare Associated with LHRH-Agonist Therapy. Rev. Urol. 2001, 3, S10–S14. [Google Scholar] [PubMed]
- Heidegger, I.; Massoner, P.; Eder, I.E.; Pircher, A.; Pichler, R.; Aigner, F.; Bektic, J.; Horninger, W.; Klocker, H. Novel therapeutic approaches for the treatment of castration-resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013, 138, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.; Boccon-Gibod, L.; Shore, N.D.; Andreou, C.; Persson, B.E.; Cantor, P.; Jensen, J.K.; Olesen, T.K.; Schroder, F.H. The efficacy and safety of degarelix: A 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int. 2008, 102, 1531–1538. [Google Scholar] [CrossRef]
- Crawford, E.D.; Hou, A.H. The role of LHRH antagonists in the treatment of prostate cancer. Oncology (Williston Park) 2009, 23, 626–630. [Google Scholar]
- Gomella, L.G. Effective testosterone suppression for prostate cancer: is there a best castration therapy? Rev. Urol. 2009, 11, 52–60. [Google Scholar]
- Boccon-Gibod, L.; van der Meulen, E.; Persson, B.E. An update on the use of gonadotropin-releasing hormone antagonists in prostate cancer. Ther. Adv. Urol. 2011, 3, 127–140. [Google Scholar] [CrossRef] [Green Version]
- McLeod, D.; Zinner, N.; Tomera, K.; Gleason, D.; Fotheringham, N.; Campion, M.; Garnick, M.B.; Abarelix Study, G. A phase 3, multicenter, open-label, randomized study of abarelix versus leuprolide acetate in men with prostate cancer. Urology 2001, 58, 756–761. [Google Scholar] [CrossRef]
- Debruyne, F.; Bhat, G.; Garnick, M.B. Abarelix for injectable suspension: first-in-class gonadotropin-releasing hormone antagonist for prostate cancer. Future Oncol. 2006, 2, 677–696. [Google Scholar] [CrossRef]
- Rick, F.G.; Block, N.L.; Schally, A.V. An update on the use of degarelix in the treatment of advanced hormone-dependent prostate cancer. Onco Targets Ther. 2013, 6, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damber, J.E.; Tammela, T.L.; Iversen, P.; Abrahamsson, P.A.; Boccon-Gibod, L.; Olesen, T.K.; van der Meulen, E.; Persson, B.E. The effect of baseline testosterone on the efficacy of degarelix and leuprolide: further insights from a 12-month, comparative, phase III study in prostate cancer patients. Urology 2012, 80, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Van Poppel, H.; Tombal, B.; de la Rosette, J.J.; Persson, B.E.; Jensen, J.K.; Kold Olesen, T. Degarelix: A novel gonadotropin-releasing hormone (GnRH) receptor blocker—Results from a 1-yr, multicentre, randomised, phase 2 dosage-finding study in the treatment of prostate cancer. Eur. Urol. 2008, 54, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zong, H.; Yan, H.; Li, N.; Zhang, Y. Degarelix versus goserelin plus bicalutamide therapy for lower urinary tract symptom relief, prostate volume reduction and quality of life improvement in men with prostate cancer: a systematic review and meta-analysis. Urol. Int. 2014, 93, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.; Auchus, R.J.; Hughes, B.A.; Cassidy, A.M.; Thompson, E.; Oommen, N.B.; Folkerd, E.; Dowsett, M.; Arlt, W.; et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab. 2012, 97, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Trachtenberg, J.; Pont, A. Ketoconazole therapy for advanced prostate cancer. Lancet 1984, 2, 433–435. [Google Scholar] [CrossRef]
- Pont, A.; Williams, P.L.; Azhar, S.; Reitz, R.E.; Bochra, C.; Smith, E.R.; Stevens, D.A. Ketoconazole blocks testosterone synthesis. Arch. Intern. Med. 1982, 142, 2137–2140. [Google Scholar] [CrossRef]
- Pont, A.; Williams, P.L.; Loose, D.S.; Feldman, D.; Reitz, R.E.; Bochra, C.; Stevens, D.A. Ketoconazole blocks adrenal steroid synthesis. Ann. Intern. Med. 1982, 97, 370–372. [Google Scholar] [CrossRef]
- Nakabayashi, M.; Xie, W.; Regan, M.M.; Jackman, D.M.; Kantoff, P.W.; Oh, W.K. Response to low-dose ketoconazole and subsequent dose escalation to high-dose ketoconazole in patients with androgen-independent prostate cancer. Cancer 2006, 107, 975–981. [Google Scholar] [CrossRef]
- Small, E.J.; Halabi, S.; Dawson, N.A.; Stadler, W.M.; Rini, B.I.; Picus, J.; Gable, P.; Torti, F.M.; Kaplan, E.; Vogelzang, N.J. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: A phase III trial (CALGB 9583). J. Clin. Oncol. 2004, 22, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Jennrich, R.; Strum, S.; Brosman, S.; Johnson, H.; Lam, R. Long-term outcome for men with androgen independent prostate cancer treated with ketoconazole and hydrocortisone. J. Urol. 2005, 173, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Pond, G.R.; Armstrong, A.J.; Galsky, M.D.; Wood, B.A.; Leopold, L.; Sonpavde, G. Efficacy of docetaxel-based chemotherapy following ketoconazole in metastatic castration-resistant prostate cancer: implications for prior therapy in clinical trials. Urol. Oncol. 2013, 31, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Kerle, D.J.; Ware, H.; Doble, A.; Dunlop, H.; Smith, C.; Allen, J.; Yeo, T.; Bloom, S.R. Objective responses to ketoconazole therapy in patients with relapsed progressive prostatic cancer. Br. J. Urol. 1986, 58, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J.; Yu, M.K.; Nguyen, S.; Mundle, S.D. Use of prednisone with abiraterone acetate in metastatic castration-resistant prostate cancer. Oncologist 2014, 19, 1231–1240. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bishop, A.C.; Alyamani, M.; Garcia, J.A.; Dreicer, R.; Bunch, D.; Liu, J.; Upadhyay, S.K.; Auchus, R.J.; Sharifi, N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 2015, 523, 347–351. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Sharifi, N.; Auchus, R.J.; Attard, G. Molecular pathways: Inhibiting steroid biosynthesis in prostate cancer. Clin. Cancer Res. 2013, 19, 3353–3359. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Garcia, J.A. Targeting the adrenal gland in castration-resistant prostate cancer: A case for orteronel, a selective CYP-17 17,20-lyase inhibitor. Curr. Oncol. Rep. 2013, 15, 105–112. [Google Scholar] [CrossRef]
- Saad, F.; Fizazi, K.; Jinga, V.; Efstathiou, E.; Fong, P.C.; Hart, L.L.; Jones, R.; McDermott, R.; Wirth, M.; Suzuki, K.; et al. Orteronel plus prednisone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (ELM-PC 4): a double-blind, multicentre, phase 3, randomised, placebo-controlled trial. Lancet Oncol. 2015, 16, 338–348. [Google Scholar] [CrossRef]
- Toren, P.J.; Kim, S.; Pham, S.; Mangalji, A.; Adomat, H.; Guns, E.S.; Zoubeidi, A.; Moore, W.; Gleave, M.E. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol. Cancer Ther. 2015, 14, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juniewicz, P.E.; McCarthy, M.; Lemp, B.M.; Barbolt, T.A.; Shaw, C.; Hollenbaugh, D.M.; Winneker, R.C.; Reel, J.R.; Batzold, F.H. The effect of the steroidal androgen receptor antagonist, Win 49,596, on the prostate and testis of beagle dogs. Endocrinology 1990, 126, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Winneker, R.C.; Wagner, M.M.; Batzold, F.H. Studies on the mechanism of action of Win 49596: A steroidal androgen receptor antagonist. J. Steroid Biochem. 1989, 33, 1133–1138. [Google Scholar] [CrossRef]
- Robinson, M.R.; Smith, P.H.; Richards, B.; Newling, D.W.; de Pauw, M.; Sylvester, R. The final analysis of the EORTC Genito-Urinary Tract Cancer Co-Operative Group phase III clinical trial (protocol 30805) comparing orchidectomy, orchidectomy plus cyproterone acetate and low dose stilboestrol in the management of metastatic carcinoma of the prostate. Eur. Urol. 1995, 28, 273–283. [Google Scholar] [PubMed]
- Prostate Cancer Trialists’ Collaborative Group. Maximum androgen blockade in advanced prostate cancer: An overview of the randomised trials. Lancet 2000, 355, 1491–1498. [Google Scholar]
- Thorpe, S.C.; Azmatullah, S.; Fellows, G.J.; Gingell, J.C.; O’Boyle, P.J. A prospective, randomised study to compare goserelin acetate (Zoladex) versus cyproterone acetate (Cyprostat) versus a combination of the two in the treatment of metastatic prostatic carcinoma. Eur. Urol. 1996, 29, 47–54. [Google Scholar] [CrossRef]
- D’Ancona, F.C.; Debruyne, F.M. Endocrine approaches in the therapy of prostate carcinoma. Hum. Reprod. Update 2005, 11, 309–317. [Google Scholar] [CrossRef]
- Osguthorpe, D.J.; Hagler, A.T. Mechanism of androgen receptor antagonism by bicalutamide in the treatment of prostate cancer. Biochemistry 2011, 50, 4105–4113. [Google Scholar] [CrossRef] [Green Version]
- Goldspiel, B.R.; Kohler, D.R. Flutamide: An antiandrogen for advanced prostate cancer. DICP 1990, 24, 616–623. [Google Scholar] [CrossRef]
- Davis, N.B.; Ryan, C.W.; Stadler, W.M.; Vogelzang, N.J. A phase II study of nilutamide in men with prostate cancer after the failure of flutamide or bicalutamide therapy. BJU Int. 2005, 96, 787–790. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathkopf, D.E.; Morris, M.J.; Fox, J.J.; Danila, D.C.; Slovin, S.F.; Hager, J.H.; Rix, P.J.; Chow Maneval, E.; Chen, I.; Gonen, M.; et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J. Clin. Oncol. 2013, 31, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- Omlin, A.; Jones, R.J.; van der Noll, R.; Satoh, T.; Niwakawa, M.; Smith, S.A.; Graham, J.; Ong, M.; Finkelman, R.D.; Schellens, J.H.; et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer—Results of two parallel first-in-human phase I studies. Investig. New Drugs 2015, 33, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Shore, N.D. Darolutamide (ODM-201) for the treatment of prostate cancer. Expert Opin. Pharmacother. 2017, 18, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Tetel, M.J. Nuclear receptor coactivators in neuroendocrine function. J. Neuroendocrinol. 2000, 12, 927–932. [Google Scholar] [CrossRef]
- Tetel, M.J. Nuclear receptor coactivators: Essential players for steroid hormone action in the brain and in behaviour. J. Neuroendocrinol. 2009, 21, 229–237. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.M.; Gao, A.C. Current strategies for targeting the activity of androgen receptor variants. Asian J. Urol. 2019, 6, 42–49. [Google Scholar] [CrossRef]
- Peacock, S.O.; Fahrenholtz, C.D.; Burnstein, K.L. Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Mol. Endocrinol. 2012, 26, 1967–1979. [Google Scholar] [CrossRef] [Green Version]
- Magani, F.; Peacock, S.O.; Rice, M.A.; Martinez, M.J.; Greene, A.M.; Magani, P.S.; Lyles, R.; Weitz, J.R.; Burnstein, K.L. Targeting AR Variant-Coactivator Interactions to Exploit Prostate Cancer Vulnerabilities. Mol. Cancer Res. 2017, 15, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.J.; Jeong, B.C.; Kim, H.J.; Lim, J.E.; Kwon, G.Y.; Kim, J.H. DBC1 promotes castration-resistant prostate cancer by positively regulating DNA binding and stability of AR-V7. Oncogene 2018, 37, 1326–1339. [Google Scholar] [CrossRef] [PubMed]
- De Mol, E.; Fenwick, R.B.; Phang, C.T.; Buzon, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; Garcia, J.; Bertoncini, C.W.; Estebanez-Perpina, E.; et al. EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverria, P.C.; Picard, D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta 2010, 1803, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Morishima, Y.; Osawa, Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J. Biol. Chem. 2008, 283, 22885–22889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, M.; Fitze, G. HSP90AB1: Helping the good and the bad. Gene 2016, 575, 171–186. [Google Scholar] [CrossRef] [Green Version]
- De Leon, J.T.; Iwai, A.; Feau, C.; Garcia, Y.; Balsiger, H.A.; Storer, C.L.; Suro, R.M.; Garza, K.M.; Lee, S.; Kim, Y.S.; et al. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11878–11883. [Google Scholar] [CrossRef] [Green Version]
- Saporita, A.J.; Ai, J.; Wang, Z. The Hsp90 inhibitor, 17-AAG, prevents the ligand-independent nuclear localization of androgen receptor in refractory prostate cancer cells. Prostate 2007, 67, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Heath, E.I.; Hillman, D.W.; Vaishampayan, U.; Sheng, S.; Sarkar, F.; Harper, F.; Gaskins, M.; Pitot, H.C.; Tan, W.; Ivy, S.P.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 2008, 14, 7940–7946. [Google Scholar] [CrossRef] [Green Version]
- Slovin, S.; Hussain, S.; Saad, F.; Garcia, J.; Picus, J.; Ferraldeschi, R.; Crespo, M.; Flohr, P.; Riisnaes, R.; Lin, C.; et al. Pharmacodynamic and Clinical Results from a Phase I/II Study of the HSP90 Inhibitor Onalespib in Combination with Abiraterone Acetate in Prostate Cancer. Clin. Cancer Res. 2019, 25, 4624–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, Y.H.; Hiipakka, R.A.; Liao, S. Modulation of endocrine systems and food intake by green tea epigallocatechin gallate. Endocrinology 2000, 141, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Hiipakka, R.A. Selective inhibition of steroid 5 alpha-reductase isozymes by tea epicatechin-3-gallate and epigallocatechin-3-gallate. Biochem. Biophys. Res. Commun. 1995, 214, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Asim, M.; Hafeez, B.B.; Adhami, V.M.; Tarapore, R.S.; Mukhtar, H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J. 2011, 25, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Fujita, R.; Liu, J.; Shimizu, K.; Konishi, F.; Noda, K.; Kumamoto, S.; Ueda, C.; Tajiri, H.; Kaneko, S.; Suimi, Y.; et al. Anti-androgenic activities of Ganoderma lucidum. J. Ethnopharmacol. 2005, 102, 107–112. [Google Scholar] [CrossRef]
- Grant, P.; Ramasamy, S. An update on plant derived anti-androgens. Int. J. Endocrinol. Metab. 2012, 10, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, H.; Xiao, Z.; Ishida, J.; Nagai, M.; Wang, H.K.; Itokawa, H.; Su, C.Y.; Shih, C.; Chiang, T.; Chang, E.; et al. Antitumor agents. 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J. Med. Chem. 2002, 45, 5037–5042. [Google Scholar] [CrossRef]
- Wei, X.; Du, Z.Y.; Zheng, X.; Cui, X.X.; Conney, A.H.; Zhang, K. Synthesis and evaluation of curcumin-related compounds for anticancer activity. Eur. J. Med. Chem. 2012, 53, 235–245. [Google Scholar] [CrossRef]
- Zhou, D.Y.; Ding, N.; Du, Z.Y.; Cui, X.X.; Wang, H.; Wei, X.C.; Conney, A.H.; Zhang, K.; Zheng, X. Curcumin analogues with high activity for inhibiting human prostate cancer cell growth and androgen receptor activation. Mol. Med. Rep. 2014, 10, 1315–1322. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [Green Version]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Soest, R.V.; Andersen, R.J. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org. Lett. 2008, 10, 4947–4950. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, H.; Hawthorn, L.; Ganther, H.E.; Ip, C. Delineation of the molecular basis for selenium-induced growth arrest in human prostate cancer cells by oligonucleotide array. Cancer Res. 2003, 63, 52–59. [Google Scholar] [PubMed]
- Christensen, M.J.; Nartey, E.T.; Hada, A.L.; Legg, R.L.; Barzee, B.R. High selenium reduces NF-kappaB-regulated gene expression in uninduced human prostate cancer cells. Nutr. Cancer 2007, 58, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Shiota, M.; Tanaka, M.; Otsuka, A.; Matsumoto, M.; Kato, M.; Tamada, S.; Iwao, H.; Miura, K.; Nakatani, T.; et al. Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCaP95 prostate cancer cells. Cancer Sci. 2017, 108, 1820–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, B.; Liu, X.; Fu, X.; Xiong, Z.; Chen, L.; Sartor, O.; Dong, Y.; Zhang, H. Berberine suppresses androgen receptor signaling in prostate cancer. Mol. Cancer Ther. 2011, 10, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med. 1999, 5, 280–285. [Google Scholar] [CrossRef]
- Franco, O.E.; Onishi, T.; Yamakawa, K.; Arima, K.; Yanagawa, M.; Sugimura, Y.; Kawamura, J. Mitogen-activated protein kinase pathway is involved in androgen-independent PSA gene expression in LNCaP cells. Prostate 2003, 56, 319–325. [Google Scholar] [CrossRef]
- Lin, D.L.; Whitney, M.C.; Yao, Z.; Keller, E.T. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin. Cancer Res. 2001, 7, 1773–1781. [Google Scholar]
- Bhardwaj, A.; Singh, S.; Srivastava, S.K.; Arora, S.; Hyde, S.J.; Andrews, J.; Grizzle, W.E.; Singh, A.P. Restoration of PPP2CA expression reverses epithelial-to-mesenchymal transition and suppresses prostate tumour growth and metastasis in an orthotopic mouse model. Br. J. Cancer 2014, 110, 2000–2010. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Singh, S.; Srivastava, S.K.; Honkanen, R.E.; Reed, E.; Singh, A.P. Modulation of protein phosphatase 2A activity alters androgen-independent growth of prostate cancer cells: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Jia, J.; Tong, Q.; Liu, J.; Qiu, J.; Sun, R.; Yao, L.; Yang, C. Knockdown of cancerous inhibitor of protein phosphatase 2A may sensitize metastatic castration-resistant prostate cancer cells to cabazitaxel chemotherapy. Tumour Biol. 2015, 36, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Arora, S.; McClellan, S.; Grizzle, W.E.; Reed, E.; Singh, A.P. Myb overexpression overrides androgen depletion-induced cell cycle arrest and apoptosis in prostate cancer cells, and confers aggressive malignant traits: potential role in castration resistance. Carcinogenesis 2012, 33, 1149–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fradet, A.; Bouchet, M.; Delliaux, C.; Gervais, M.; Kan, C.; Benetollo, C.; Pantano, F.; Vargas, G.; Bouazza, L.; Croset, M.; et al. Estrogen related receptor alpha in castration-resistant prostate cancer cells promotes tumor progression in bone. Oncotarget 2016, 7, 77071–77086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonkhoff, H. Estrogen receptor signaling in prostate cancer: Implications for carcinogenesis and tumor progression. Prostate 2018, 78, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Q. The role of glucocorticoid receptor in prostate cancer progression: From bench to bedside. Int. Urol. Nephrol. 2017, 49, 369–380. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| LHRH Agonist | ||
|---|---|---|
| Compound | Mechanism of Action | Ref |
| Buserelin * | Binds to and desensitizes LHRH receptor, and thus, reduces the level of testosterone. Not available in US, but marketed elsewhere in the world | [84,85] |
| Leuprolide * | Binds to LHRH receptor and acts as an inhibitor of gonadotropin secretion. The prolonged exposure of the agonist decreases the secretion of LH, FSH, and testosterone. | [86] |
| Goserelin * | Binds to and activates the LHRH receptor to inhibit the release of pituitary gonadotropin and thus reduces the testosterone level. | [87,88] |
| Histrelin * | Binds to LHRH receptor and acts as an inhibitor of gonadotropin secretion. The continuous administration of this agonist reduces the levels of LH, FSH and testosterone. This is marketed as Vantas. | [89,90] |
| Triptorelin * | Binds to LHRH receptor and whose prolonged exposure is shown to decrease the secretion of LH, FSH and testosterone. | [91,92] |
| LHRH Antagonist | ||
| Abarelix * | Binds to LHRH receptor and acts as a potent inhibitor without initial testosterone surge. Currently available in Germany, but was withdrawn in the US in 2005. | [93] |
| Cetrorelix ** | Binds to the LHRH receptor and inhibits the secretion of LH and FSH. Not yet approved for prostate cancer, but indicated for the inhibition of premature LH surges in women undergoing controlled ovarian stimulation. | [94] |
| Ganirelix ** | Binds to and prevents the LHRH receptor from LHRH binding (with no initial testosterone surge) and thus reducing the release of gonadotropin and testosterone. Not yet approved for prostate cancer, but indicated for the inhibition of premature LH surges in women undergoing controlled ovarian stimulation | [95] |
| Degarelix * | Binds to and prevents the LHRH receptor from LHRH binding (with no initial testosterone surge) thus reducing the release of LH, FSH, and testosterone. | [96] |
| Androgen Synthesis Inhibitor | ||
| Abiraterone * | Covalently binds to and selectively inhibits the androgen biosynthesis enzyme, CYP17A, in an irreversible manner and hence, reduces the level of testosterone and other androgens. | [97] |
| Orteronel (TAK-700) * | Inhibits the androgen biosynthesis enzyme, CYP17A, and thus reduces the level of testosterone. | [98] |
| Finasteride * | Inhibits the synthesis of DHT from testosterone. | [99] |
| Anti-Androgen: Steroidal | ||
|---|---|---|
| Compound | Mechanism of Action | Ref |
| Cyproterone acetate * | Not only functions as an anti-androgen but also possesses potent anti-gonadotropic activity that results in rapid suppression of serum testosterone. The use in clinics has been discontinued | [100] |
| Megestrol acetate * | Exerts its effects through various mechanisms. Primarily, it acts as an anti-androgen, but can also inhibit 5-alpha reductase and LH release. | [101] |
| Dienogest ** | Binds to and blocks the binding of androgens to the androgen receptor | [102] |
| Galeterone (TOK-001) * | It possesses a dual mechanism of action, acting as both as an anti-androgen and as a CYP17A1 inhibitor suppressing the biosynthesis of androgen | [103] |
| Chlormadinone acetate * | Acts as a partial antagonist of AR. Also reduces the activity of 5α-reductase and thus inhibiting androgen production and signaling Not approved in US. | [104] |
| Anti-Androgen: Non-Steroidal | ||
| First Generation Inhibitor | ||
| Bicalutamide * | Binds to the allosteric site on the AR, induces a conformational change in the co-activator binding site and thus interferes with its transcriptional activity | [105] |
| Flutamide * | Competitively binds to AR and inhibits the binding of androgen to AR. | [106] |
| Nilutamide * | Competitively binds to AR and inhibits the binding of androgen to AR. | [107] |
| Second Generation Inhibitor | ||
| Enzalutamide (MDV 3100) * | Selectively binds to AR with high affinity and blocks the nuclear translocation of AR, inhibits the recruitment of coactivator and AR DNA binding. | [108] |
| Apalutamide (ARN-509) * | Competitively binds to AR with high affinity, reduces the binding of androgen to AR, and inhibits the nuclear translocation of AR. | [109] |
| AZD3514 ** | Binds to the LBD of AR and thus inhibits the ligand-driven nuclear AR translocation. | [110] |
| Darolutamide (ODM201) * | Binds to AR with high affinity and reduces the binding of androgen to AR leading to inhibition of the nuclear translocation of AR. Some consider it as a “third generation anti-androgen” as its potency is not affected by the F876L AR mutation that is considered critical for the resistance to Enzalutamide and Apalutamide. | [111] |
| Targeting the Binding of AR and Co-Regulators | ||
|---|---|---|
| Compound | Mechanism of Action | Ref |
| MCB-613 ** | The inhibitor of NTD that interacts with AF-1 region and inhibits AR activation and AR-mediated signaling pathway. | [112] |
| EPI-001 *** | Selectively binds AF-1 domain of the androgen receptor and thus represses the transcriptional activity of AR. | [113] |
| Ralaniten (EPI-506) ** | The derivative of EPI-001, which acts as an inhibitor of NTD that interact with AF-1 region leading to the inhibition of AR activation and AR-mediated signaling pathway. | [114] |
| EPI-002 ** | The stereoisomer of EPI-001, which has the potency to disrupt the NTD of AR and inhibits the transcriptional activity of AR. | [115] |
| Targeting AR Co-Factor(s) | ||
| 17-AAG *** | Inhibits HSP90 and the ligand-independent nuclear localization of AR | [116] |
| Methoxychalcones ** | Stabilizes AR-Heat shock protein complex and thus prevents AR dimerization. | [117] |
| Onalespib * | Inhibits HSP90 leading to the degradation of client proteins including AR. | [118] |
| Compound | Mechanism of Action | Ref |
|---|---|---|
| Green tea extract (EGCG) | The polyphenol in green tea extract inhibits 5α-reductase activity thus impedes the androgen synthesis. | [119] |
| Mushroom extract | The triterpenoid compound in the mushroom extract inhibits 5α-reductase activity and thus impedes the androgen synthesis. | [120] |
| Glycyrrhetinic acid | Targets 17,20-lyase enzyme and thus decreases the testosterone level. | [121] |
| Curcumin | Curcumin analogs function as androgen antagonists, inhibit testosterone-, DHT-induced AR activity. | [122] |
| Niphatenones | The glycerol ethers from the sponge Niphates digitalis, which covalently binds to the AF1 region of the NTD. | [123] |
| Sintokamides | The derivative of the marine sponge Dysidea sp that binds to N-terminal domain of AR and thus, suppressed the AR activity. | [124] |
| Selenium | Regulates AR gene expression. | [125] |
| Quercetin | The flavonol pigment in onion and apple acts as the HSP70 inhibitor and induces the AR degradation. | [126] |
| Berberine | The plant phytochemical, which acts as the HSP70 inhibitor and thus induces the AR degradation. | [127] |
| Hydroxytyrosol | Inhibits AR expression. | [128] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers 2020, 12, 51. https://doi.org/10.3390/cancers12010051
Saranyutanon S, Srivastava SK, Pai S, Singh S, Singh AP. Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers. 2020; 12(1):51. https://doi.org/10.3390/cancers12010051
Chicago/Turabian StyleSaranyutanon, Sirin, Sanjeev Kumar Srivastava, Sachin Pai, Seema Singh, and Ajay Pratap Singh. 2020. "Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope" Cancers 12, no. 1: 51. https://doi.org/10.3390/cancers12010051
APA StyleSaranyutanon, S., Srivastava, S. K., Pai, S., Singh, S., & Singh, A. P. (2020). Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers, 12(1), 51. https://doi.org/10.3390/cancers12010051