Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
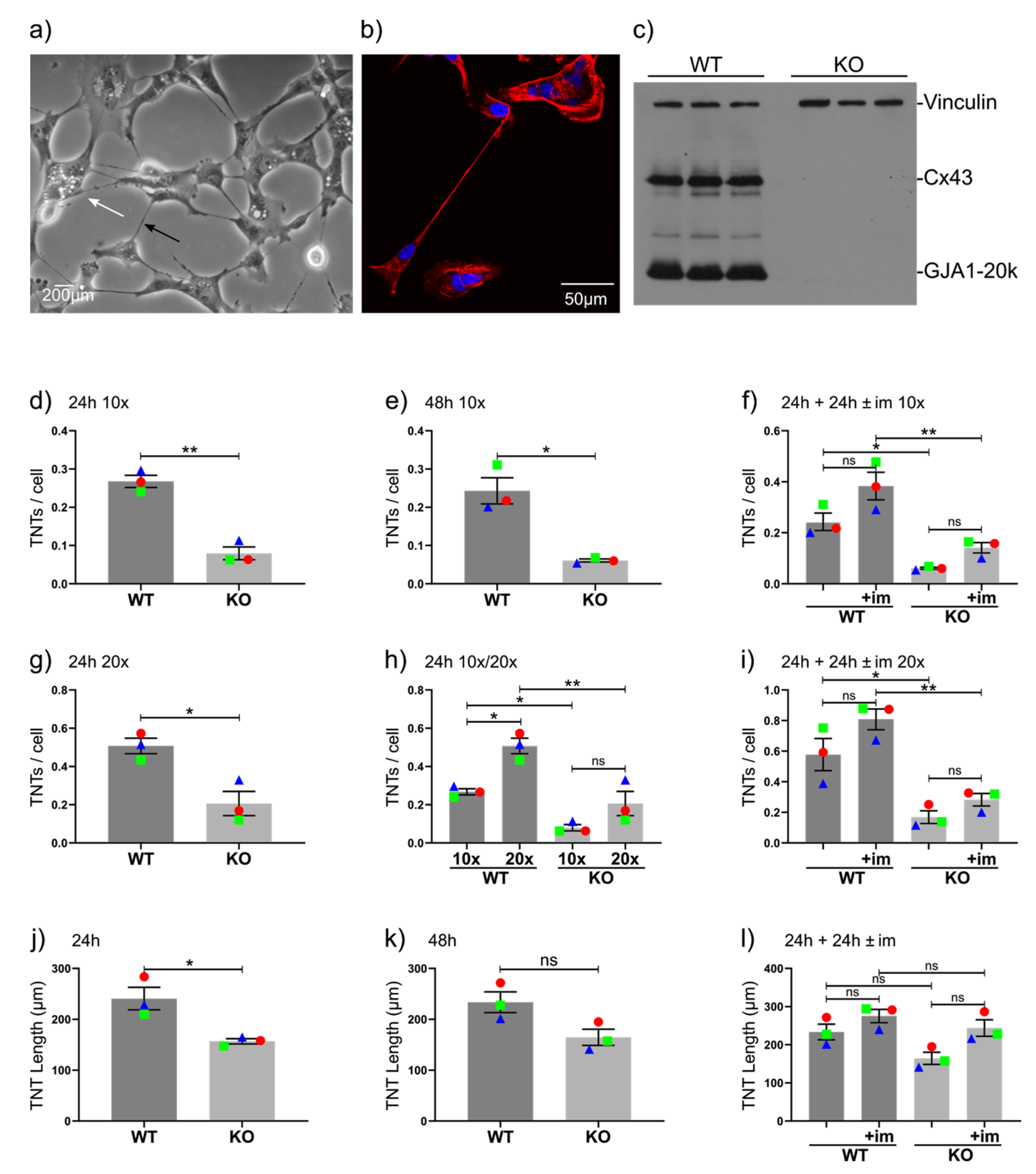
2.1. GJA1 Knockout Reduces TNT Number and Length in BT549 Breast Cancer Cells
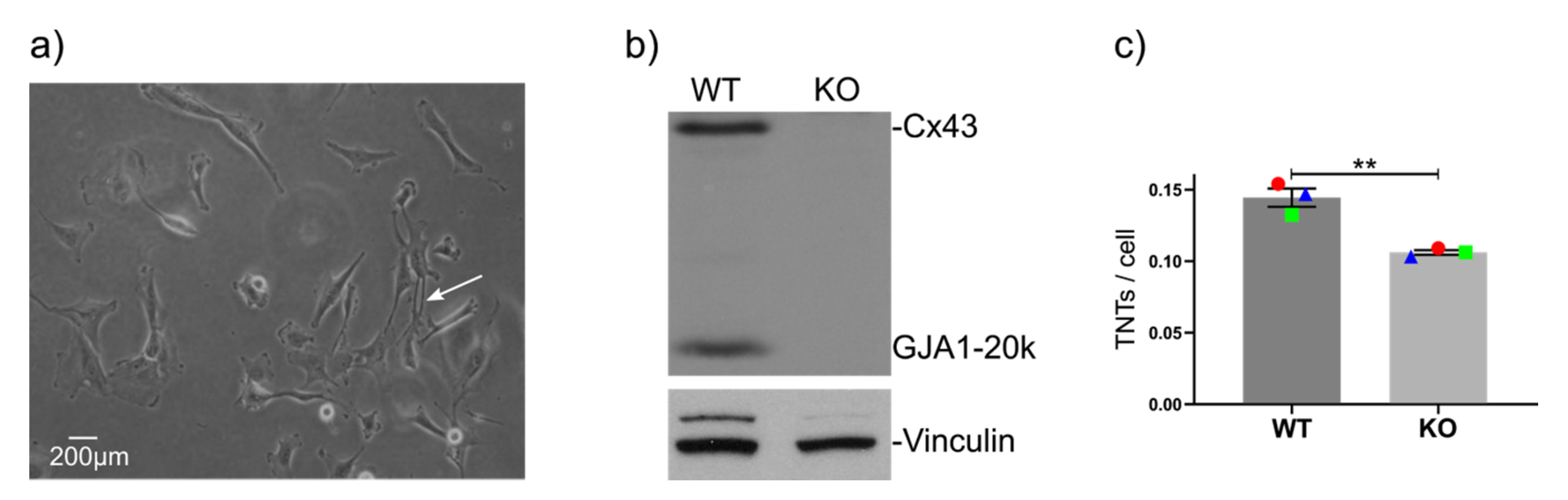
2.2. GJA1 Knockout Reduces the Number of TNTs in Hs578t Breast Cancer Cells
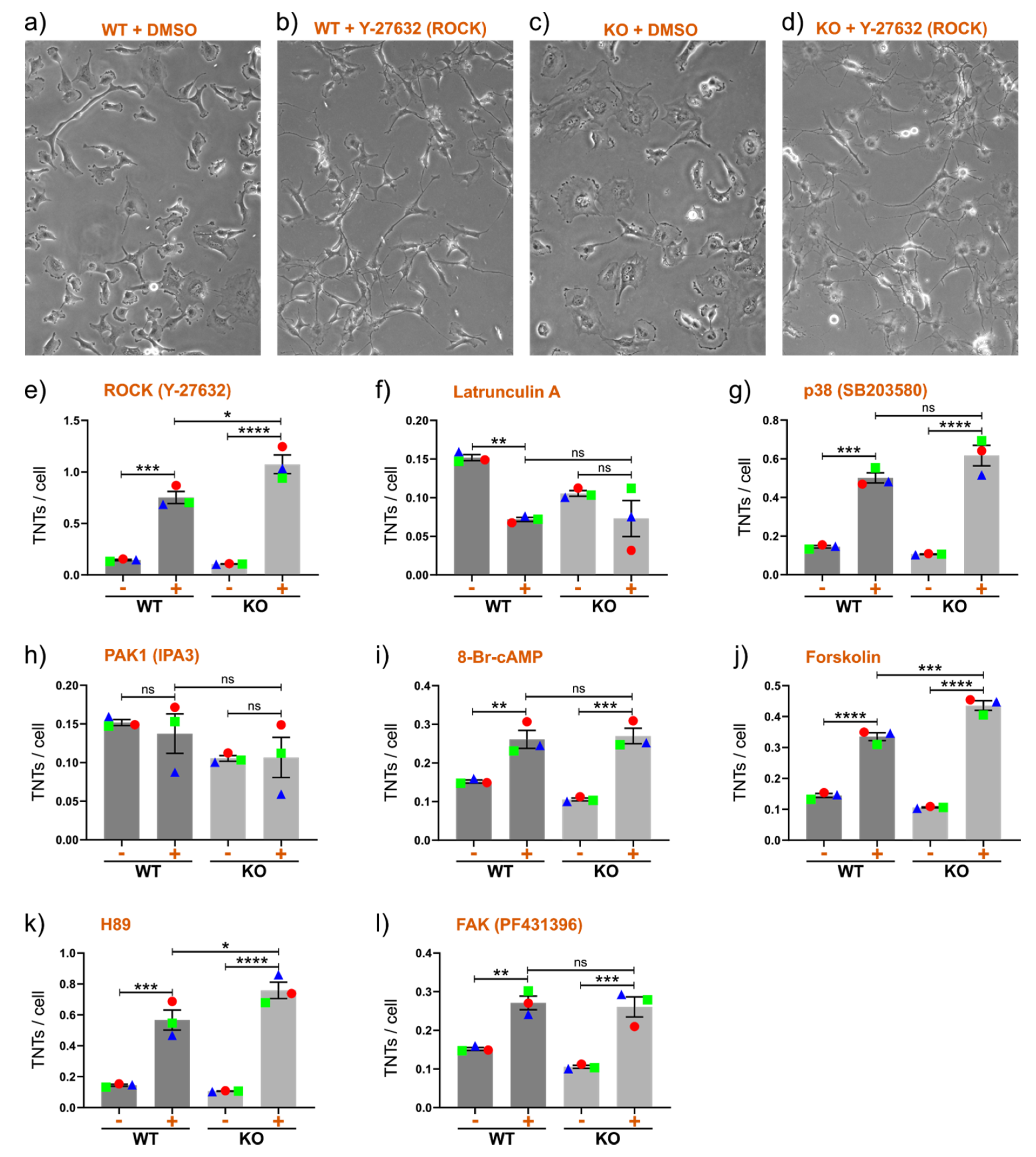
2.3. Signaling Pathways Linked to Cx43 Differentially Affect TNT Formation in Cx43 WT and KO Cells
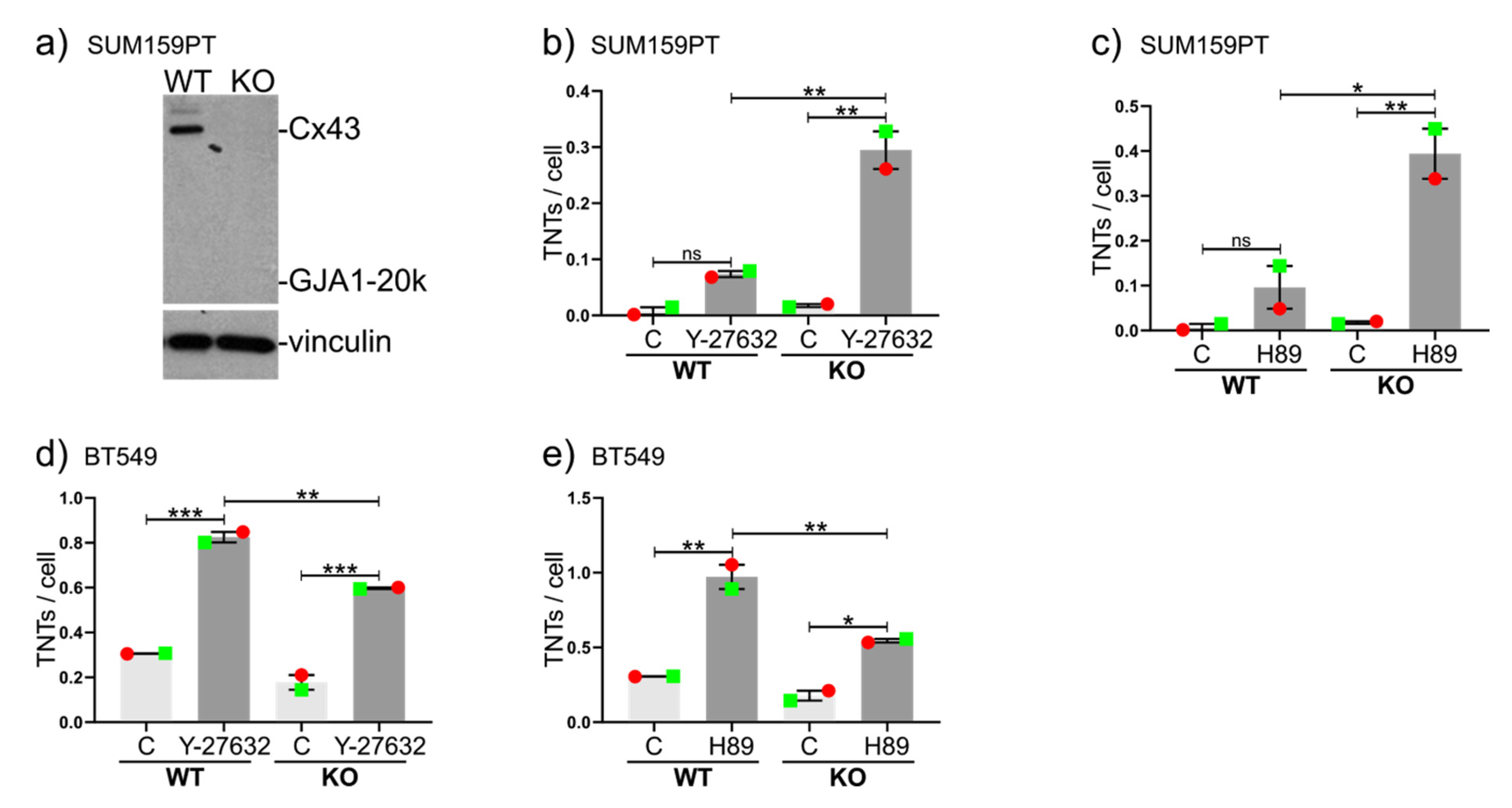
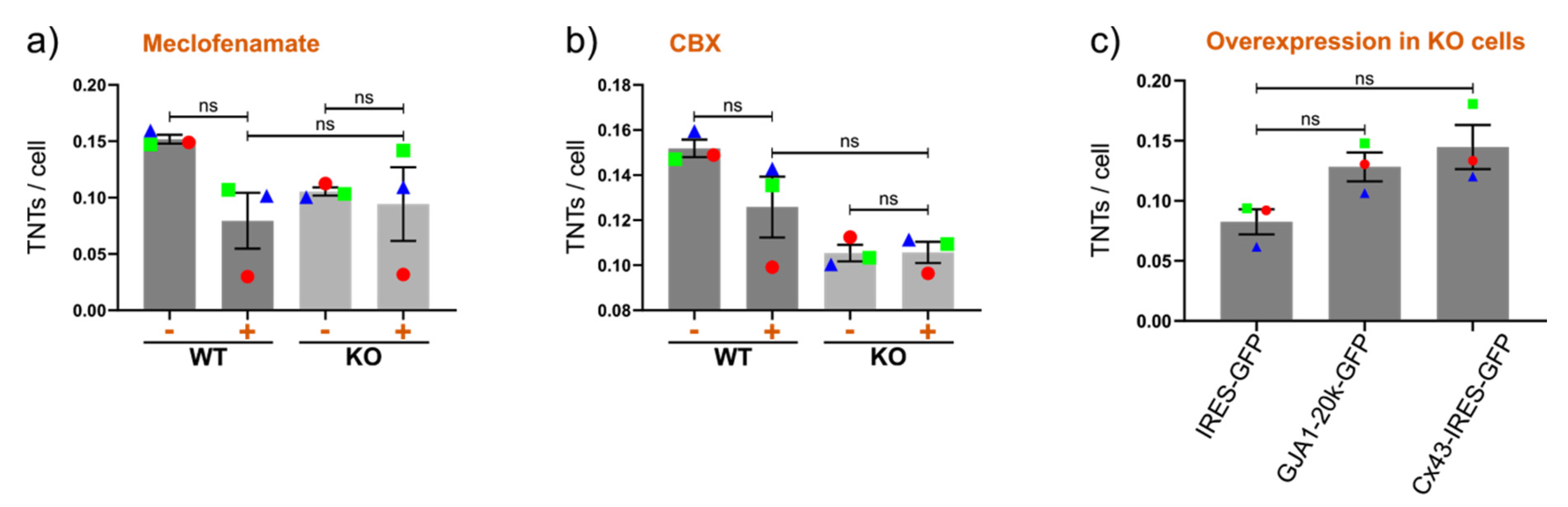
2.4. Drug-Induced Stimulation of TNTs in the Presence or Absence of Cx43 in Cell Lines Expressing Very High or Very Low Numbers of TNTs under Normal Conditions
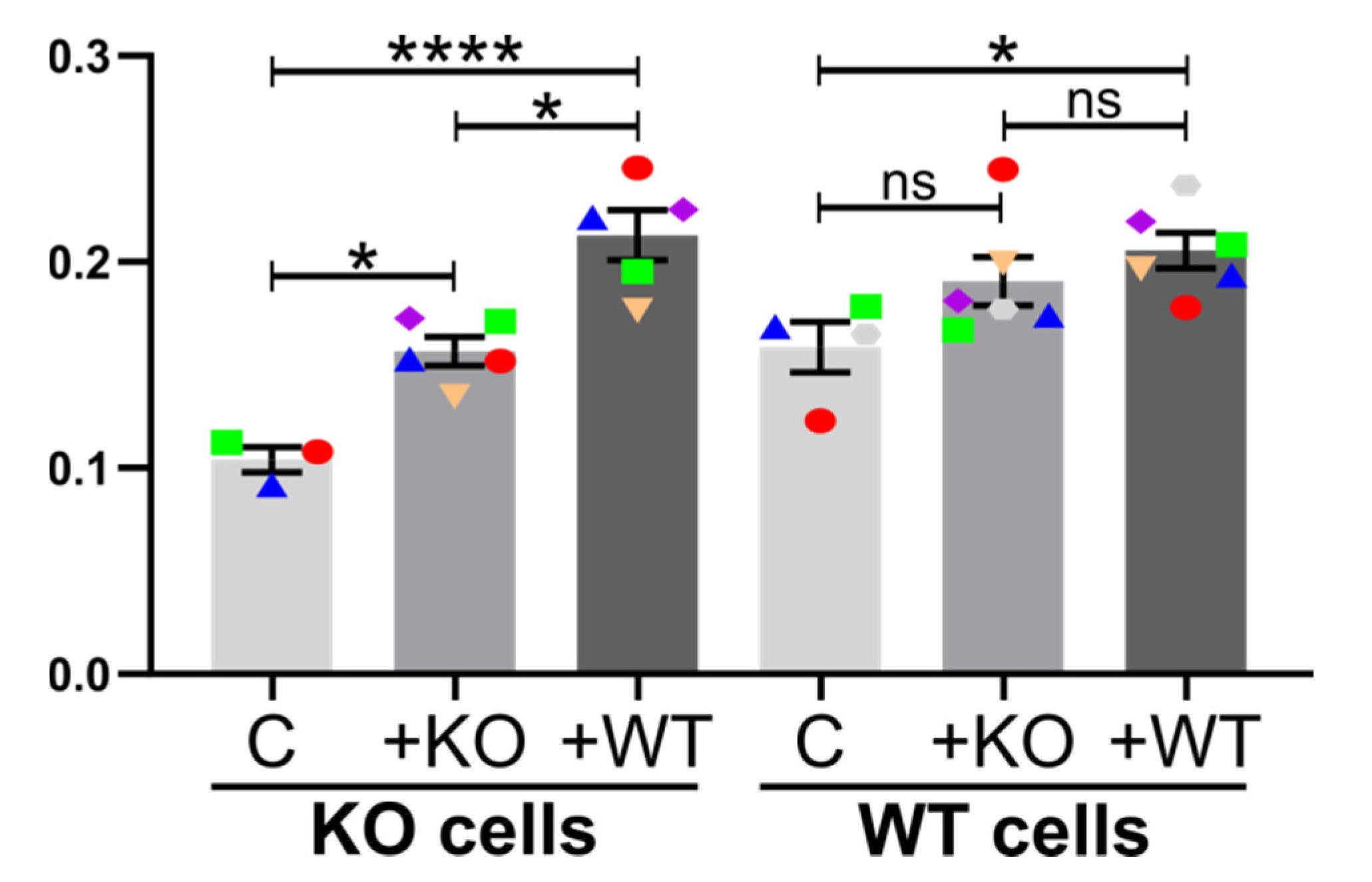
2.5. Further Insight into Cx43 Pathways Stimulating TNT Formation
2.5.1. The role of GJIC
2.5.2. Channel-Independent Effects
2.5.3. Induction of TNTs by Conditioned Media
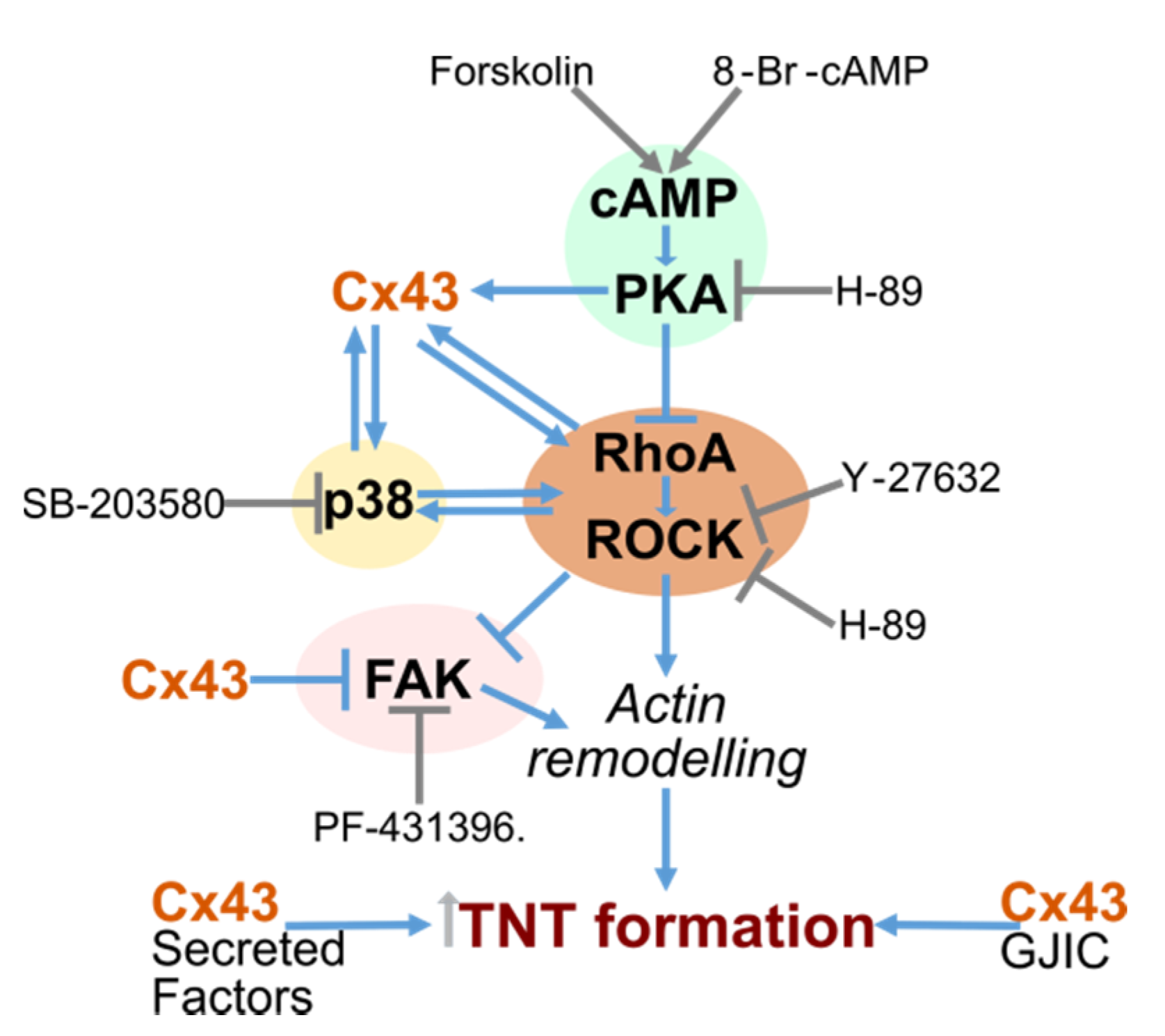
3. Discussion
4. Materials and Methods
4.1. Cx43 Knockout and Overexpression of Cx43 and GJA1-20k
4.1.1. Generation of an All-in-One High-Fidelity Cbh-hfCas9-2A-eGFP CRISPR Plasmid
4.1.2. Cbh-hfCas9-2A-eGFP CRISPR System Targeting Cx43
4.1.3. Generation of KO Clones
4.1.4. Overexpression of Cx43 and GJA1-20k
4.2. Cell Culture
4.3. TNT Analysis
4.4. Drug Treatments
4.5. Peptide Treatments
4.6. Conditioned Medium Treatment
4.7. Protein Extraction and Quantification
4.8. Western Blotting
4.9. Immunofluorescence Microscopy
4.10. F-actin/G-actin Ratio Determination
4.11. Parachute Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T.W. Connexins and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesnil, M.; Aasen, T.; Boucher, J.; Chépied, A.; Cronier, L.; Defamie, N.; Kameritsch, P.; Laird, D.W.; Lampe, P.D.; Lathia, J.D.; et al. An update on minding the gap in cancer. Biochim. Biophys. Acta Biomembr. 2018, 1860, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological Functions of Connexin43 Beyond Intercellular Communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Anjo, S.I.; Manadas, B.; Sluijter, J.P.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins-Marques, T.; Pinho, M.J.; Zuzarte, M.; Oliveira, C.; Pereira, P.; Sluijter, J.P.G.; Gomes, C.; Girao, H. Presence of Cx43 in extracellular vesicles reduces the cardiotoxicity of the anti-tumour therapeutic approach with doxorubicin. J. Extracell Vesicles 2016, 5, 32538. [Google Scholar] [CrossRef]
- Salat-Canela, C.; Sesé, M.; Peula, C.; Ramón y Cajal, S.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. 2014, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ul-Hussain, M.; Olk, S.; Schoenebeck, B.; Wasielewski, B.; Meier, C.; Prochnow, N.; May, C.; Galozzi, S.; Marcus, K.; Zoidl, G.; et al. Internal ribosomal entry site (IRES) activity generates endogenous carboxyl-terminal domains of Cx43 and is responsive to hypoxic conditions. J. Biol. Chem. 2014, 289, 20979–20990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basheer, W.A.; Fu, Y.; Shimura, D.; Xiao, S.; Agvanian, S.; Hernandez, D.M.; Hitzeman, T.C.; Hong, T.; Shaw, R.M. Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Zhang, S.-S.; Xiao, S.; Basheer, W.A.; Baum, R.; Epifantseva, I.; Hong, T.; Shaw, R.M. Cx43 Isoform GJA1-20k Promotes Microtubule Dependent Mitochondrial Transport. Front. Physiol. 2017, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, P.; Hong, T.; Wang, Y.; Liu, N.; He, B.; Zou, S.; Ren, D.; Duan, J.; Zhao, L.; et al. Astrocytes-derived exosomes induce neuronal recovery after traumatic brain injury via delivering gap junction alpha 1-20 k. J. Tissue Eng. Regen. Med. 2019. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions-Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef]
- Mittal, R.; Karhu, E.; Wang, J.-S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell. Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.C.; Salter, R.D. Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity 2005, 23, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Thayanithy, V.; Dickson, E.L.; Steer, C.; Subramanian, S.; Lou, E. Tumor-stromal cross talk: Direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl. Res. 2014, 164, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biran, A.; Perelmutter, M.; Gal, H.; Burton, D.G.A.; Ovadya, Y.; Vadai, E.; Geiger, T.; Krizhanovsky, V. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015, 29, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Victoria, G.S.; Marzo, L.; Ghosh, R.; Zurzolo, C. Prion aggregates transfer through tunneling nanotubes in endocytic vesicles. Prion 2015, 9, 125–135. [Google Scholar] [CrossRef]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Eugenin, E.A.; Gaskill, P.J.; Berman, J.W. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell. Immunol. 2009, 254, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Jansens, R.J.J.; Tishchenko, A.; Favoreel, H.W. Bridging the Gap: Virus Long-Distance Spread via Tunneling Nanotubes. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Kim, B.-W.; Lee, J.-S.; Ko, Y.-G. Mycoplasma exploits mammalian tunneling nanotubes for cell-to-cell dissemination. BMB Rep. 2019, 52, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Koyanagi, M.; Brandes, R.P.; Haendeler, J.; Zeiher, A.M.; Dimmeler, S. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: A novel mechanism for cell fate changes? Circ. Res. 2005, 96, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.K.P.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.A.; French, P.M.W.; Davis, D.M. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [Green Version]
- Austefjord, M.W.; Gerdes, H.-H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef]
- Onfelt, B.; Nedvetzki, S.; Yanagi, K.; Davis, D.M. Cutting edge: Membrane nanotubes connect immune cells. J. Immunol. 2004, 173, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osswald, M.; Jung, E.; Wick, W.; Winkler, F. Tunneling nanotube-like structures in brain tumors. Cancer Rep. 2019, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Matejka, N.; Reindl, J. Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects. Radiat. Oncol. 2019, 14, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef] [Green Version]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A.S. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Desir, S.; O’Hare, P.; Vogel, R.I.; Sperduto, W.; Sarkari, A.; Dickson, E.L.; Wong, P.; Nelson, A.C.; Fong, Y.; Steer, C.J.; et al. Chemotherapy-Induced Tunneling Nanotubes Mediate Intercellular Drug Efflux in Pancreatic Cancer. Sci. Rep. 2018, 8, 9484. [Google Scholar] [CrossRef] [PubMed]
- Lou, E. Intercellular conduits in tumours: The new social network. Trends Cancer Res. 2016, 2, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, P.; Jena, S.R.; Samanta, L. Tunneling Nanotubes: A Versatile Target for Cancer Therapy. Curr. Cancer Drug Targets 2018, 18, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Lou, E.; Baldoni, J.; Okafo, G.; Eugenin, E. The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis. Int. J. Mol. Sci. 2018, 19, 1270. [Google Scholar] [CrossRef] [Green Version]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hänggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro. Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.-H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girão, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [Green Version]
- Lock, J.T.; Parker, I.; Smith, I.F. Communication of Ca(2+) signals via tunneling membrane nanotubes is mediated by transmission of inositol trisphosphate through gap junctions. Cell Calcium 2016, 60, 266–272. [Google Scholar] [CrossRef] [Green Version]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef]
- Li, X. Gap junction protein connexin43 and tunneling nanotubes in human trabecular meshwork cells. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 212–219. [Google Scholar]
- Yao, Y.; Fan, X.-L.; Jiang, D.; Zhang, Y.; Li, X.; Xu, Z.-B.; Fang, S.-B.; Chiu, S.; Tse, H.-F.; Lian, Q.; et al. Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Rep. 2018, 11, 1120–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fykerud, T.A.; Knudsen, L.M.; Totland, M.Z.; Sørensen, V.; Dahal-Koirala, S.; Lothe, R.A.; Brech, A.; Leithe, E. Mitotic cells form actin-based bridges with adjacent cells to provide intercellular communication during rounding. Cell Cycle 2016, 15, 2943–2957. [Google Scholar] [CrossRef] [PubMed]
- Activating, A. Brain Tumors Feed off Healthy Neurons. Nature 2019, 573, 539–545. [Google Scholar]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different modalities of intercellular membrane exchanges mediate cell-to-cell p-glycoprotein transfers in MCF-7 breast cancer cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef] [Green Version]
- Carter, K.P.; Hanna, S.; Genna, A.; Lewis, D.; Segall, J.E.; Cox, D. Macrophages enhance 3D invasion in a breast cancer cell line by induction of tumor cell tunneling nanotubes. Cancer Rep. 2019, 2. [Google Scholar] [CrossRef]
- Hanna, S.J.; McCoy-Simandle, K.; Leung, E.; Genna, A.; Condeelis, J.; Cox, D. Tunneling nanotubes, a novel mode of tumor cell-macrophage communication in tumor cell invasion. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Patheja, P.; Sahu, K. Macrophage conditioned medium induced cellular network formation in MCF-7 cells through enhanced tunneling nanotube formation and tunneling nanotube mediated release of viable cytoplasmic fragments. Exp. Cell Res. 2017, 355, 182–193. [Google Scholar] [CrossRef]
- McLachlan, E.; Shao, Q.; Laird, D.W. Connexins and gap junctions in mammary gland development and breast cancer progression. J. Membr. Biol. 2007, 218, 107–121. [Google Scholar] [CrossRef]
- Sinha, G.; Ferrer, A.I.; Moore, C.A.; Naaldijk, Y.; Rameshwar, P. Gap Junctions and Breast Cancer Dormancy. Trends Cancer Res. 2020, 6, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.L.; Williams, C.B.; Zambrano, J.N.; Williams, C.J.; Yeh, E.S. Connexin 43 in the development and progression of breast cancer: What’s the connection? (Review). Int. J. Oncol. 2017, 51, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grek, C.L.; Rhett, J.M.; Bruce, J.S.; Ghatnekar, G.S.; Yeh, E.S. Connexin 43, breast cancer tumor suppressor: Missed connections? Cancer Lett. 2016, 374, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busby, M.; Hallett, M.T.; Plante, I. The Complex Subtype-Dependent Role of Connexin 43 (GJA1) in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 693. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Wang, H.; McLachlan, E.; Veitch, G.I.L.; Laird, D.W. Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res. 2005, 65, 2705–2711. [Google Scholar] [CrossRef] [Green Version]
- Keller, K.E.; Bradley, J.M.; Sun, Y.Y.; Yang, Y.-F.; Acott, T.S. Tunneling Nanotubes are Novel Cellular Structures That Communicate Signals Between Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5298–5307. [Google Scholar] [CrossRef] [Green Version]
- Hanna, S.J.; McCoy-Simandle, K.; Miskolci, V.; Guo, P.; Cammer, M.; Hodgson, L.; Cox, D. The Role of Rho-GTPases and actin polymerization during Macrophage Tunneling Nanotube Biogenesis. Sci. Rep. 2017, 7, 8547. [Google Scholar] [CrossRef]
- Fostok, S.; El-Sibai, M.; Bazzoun, D.; Lelièvre, S.; Talhouk, R. Connexin 43 Loss Triggers Cell Cycle Entry and Invasion in Non-Neoplastic Breast Epithelium: A Role for Noncanonical Wnt Signaling. Cancers 2019, 11, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Chen, C.; Huang, K.; Wang, S.; Hao, J.; Huang, J.; Huang, H. RhoA/rho kinase signaling reduces connexin43 expression in high glucose-treated glomerular mesangial cells with zonula occludens-1 involvement. Exp. Cell Res. 2014, 327, 276–286. [Google Scholar] [CrossRef]
- Derangeon, M.; Bourmeyster, N.; Plaisance, I.; Pinet-Charvet, C.; Chen, Q.; Duthe, F.; Popoff, M.R.; Sarrouilhe, D.; Hervé, J.-C. RhoA GTPase and F-actin dynamically regulate the permeability of Cx43-made channels in rat cardiac myocytes. J. Biol. Chem. 2008, 283, 30754–30765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponsaerts, R.; D’hondt, C.; Hertens, F.; Parys, J.B.; Leybaert, L.; Vereecke, J.; Himpens, B.; Bultynck, G. RhoA GTPase switch controls Cx43-hemichannel activity through the contractile system. PLoS ONE 2012, 7, e42074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, E.; Yamanaka, H.; Kobayashi, K.; Yagi, H.; Sakagami, M.; Noguchi, K. RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. Glia 2015, 63, 216–228. [Google Scholar] [CrossRef]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. Biochim. Biophys. Acta 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [Green Version]
- Coniglio, S.J.; Zavarella, S.; Symons, M.H. Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol. Cell. Biol. 2008, 28, 4162–4172. [Google Scholar] [CrossRef] [Green Version]
- Delage, E.; Cervantes, D.C.; Pénard, E.; Schmitt, C.; Syan, S.; Disanza, A.; Scita, G.; Zurzolo, C. Differential identity of Filopodia and Tunneling Nanotubes revealed by the opposite functions of actin regulatory complexes. Sci. Rep. 2016, 6, 39632. [Google Scholar] [CrossRef]
- Howe, A.K. Regulation of actin-based cell migration by cAMP/PKA. Biochim. Biophys. Acta 2004, 1692, 159–174. [Google Scholar] [CrossRef]
- Gerits, N.; Mikalsen, T.; Kostenko, S.; Shiryaev, A.; Johannessen, M.; Moens, U. Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. J. Biol. Chem. 2007, 282, 37232–37243. [Google Scholar] [CrossRef] [Green Version]
- Pidoux, G.; Taskén, K. Anchored PKA as a gatekeeper for gap junctions. Commun. Integr. Biol. 2015, 8, e1057361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, J.M.; Phipps, M.W.; Chen, J.; Phipps, J. Cellular sublocalization of Cx43 and the establishment of functional coupling in IMR-32 neuroblastoma cells. Mol. Carcinog. 2005, 42, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Huang, F.; Lum, H. PKA inhibits RhoA activation: A protection mechanism against endothelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L972–L980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemhuis, J.; Boutillier, S.; Schmidt, G.; Meyer, D.K. The protein kinase A inhibitor H89 acts on cell morphology by inhibiting Rho kinase. J. Pharmacol. Exp. Ther. 2002, 300, 1000–1007. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H.J. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Costa, P.; Scales, T.M.E.; Ivaska, J.; Parsons, M. Integrin-specific control of focal adhesion kinase and RhoA regulates membrane protrusion and invasion. PLoS ONE 2013, 8, e74659. [Google Scholar] [CrossRef]
- Jaraíz-Rodríguez, M.; Tabernero, M.D.; González-Tablas, M.; Otero, A.; Orfao, A.; Medina, J.M.; Tabernero, A. A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Rep. 2017, 9, 451–463. [Google Scholar] [CrossRef]
- Matsuuchi, L.; Naus, C.C. Gap junction proteins on the move: Connexins, the cytoskeleton and migration. Biochim. Biophys. Acta 2013, 1828, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Aftab, Q.; Mesnil, M.; Ojefua, E.; Poole, A.; Noordenbos, J.; Strale, P.-O.; Sitko, C.; Le, C.; Stoynov, N.; Foster, L.J.; et al. Cx43-Associated Secretome and Interactome Reveal Synergistic Mechanisms for Glioma Migration and MMP3 Activation. Front. Neurosci. 2019, 13, 143. [Google Scholar] [CrossRef] [Green Version]
- Belliveau, D.J.; Bani-Yaghoub, M.; McGirr, B.; Naus, C.C.G.; Rushlow, W.J. Enhanced neurite outgrowth in PC12 cells mediated by connexin hemichannels and ATP. J. Biol. Chem. 2006, 281, 20920–20931. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bukoreshtliev, N.V.; Gerdes, H.-H. Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS ONE 2012, 7, e47429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.; Reimann, A.; Fritz, R.D.; Ryu, H.; Jeon, N.L.; Pertz, O. Spatio-temporal co-ordination of RhoA, Rac1 and Cdc42 activation during prototypical edge protrusion and retraction dynamics. Sci. Rep. 2016, 6, 21901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osteikoetxea-Molnár, A.; Szabó-Meleg, E.; Tóth, E.A.; Oszvald, Á.; Izsépi, E.; Kremlitzka, M.; Biri, B.; Nyitray, L.; Bozó, T.; Németh, P.; et al. The growth determinants and transport properties of tunneling nanotube networks between B lymphocytes. Cell. Mol. Life Sci. 2016, 73, 4531–4545. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-Y.; Li, C.-Y.; Chen, J.; Pan, L.; Saito, S.; Terashita, T.; Saito, K.; Miyawaki, K.; Shigemoto, K.; Mominoki, K.; et al. Rho-ROCK signal pathway regulates microtubule-based process formation of cultured podocytes--inhibition of ROCK promoted process elongation. Nephron Exp. Nephrol. 2004, 97, e49–e61. [Google Scholar] [CrossRef]
- Aburima, A.; Wraith, K.S.; Raslan, Z.; Law, R.; Magwenzi, S.; Naseem, K.M. cAMP signaling regulates platelet myosin light chain (MLC) phosphorylation and shape change through targeting the RhoA-Rho kinase-MLC phosphatase signaling pathway. Blood 2013, 122, 3533–3545. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.C.; Stone, C.; Tkach, L.; SundarRaj, N. Rho and Rho-kinase (ROCK) signaling in adherens and gap junction assembly in corneal epithelium. Investig. Ophthalmol. Vis. Sci. 2002, 43, 978–986. [Google Scholar]
- Suh, H.N.; Kim, M.O.; Han, H.J. Laminin-111 stimulates proliferation of mouse embryonic stem cells through a reduction of gap junctional intercellular communication via RhoA-mediated Cx43 phosphorylation and dissociation of Cx43/ZO-1/drebrin complex. Stem Cells Dev. 2012, 21, 2058–2070. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Kim, M.-J.; Mostafa, M.N.; Park, J.-H.; Choi, H.-S.; Kim, Y.-S.; Choi, E.-K. RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. Int. J. Mol. Sci. 2020, 21, 1255. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Naranjo, A.; Cormie, P.; Serrano, A.E.; Hu, R.; O’Neill, S.; Wang, C.M.; Thrasivoulou, C.; Power, K.T.; White, A.; Serena, T.; et al. Targeting Cx43 and N-cadherin, which are abnormally upregulated in venous leg ulcers, influences migration, adhesion and activation of Rho GTPases. PLoS ONE 2012, 7, e37374. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Vadnais, J.; Lu, M.L.; Noël, J.; Nabi, I.R. Rho/ROCK-dependent pseudopodial protrusion and cellular blebbing are regulated by p38 MAPK in tumour cells exhibiting autocrine c-Met activation. Biol. Cell 2006, 98, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Damodaran, N.; Dilna, A.; Kielkopf, C.S.; Kagedal, K.; Ollinger, K.; Nath, S. Amyloid-β induced membrane damage instigates tunneling nanotubes by exploiting PAK1 dependent actin remodulation. bioRxiv 2020, 655340. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Perides, G.; Wang, D.; Liu, Y.F. beta-Amyloid peptide induces formation of actin stress fibers through p38 mitogen-activated protein kinase. J. Neurochem. 2002, 83, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.B.; Robson, C.N.; Neal, D.E.; Leung, H.Y. Keratinocyte growth factor activates p38 MAPK to induce stress fibre formation in human prostate DU145 cells. Oncogene 2001, 20, 5359–5365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K. Activation of Rho-kinase and focal adhesion kinase regulates the organization of stress fibers and focal adhesions in the central part of fibroblasts. PeerJ 2017, 5, e4063. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; Xu, X.; Park, H.; Wei, C.-J.; Chang, S.; Chatterjee, B.; Lo, C. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS ONE 2011, 6, e26379. [Google Scholar] [CrossRef] [Green Version]
- Sáenz-de-Santa-María, I.; Bernardo-Castiñeira, C.; Enciso, E.; García-Moreno, I.; Chiara, J.L.; Suarez, C.; Chiara, M.-D. Control of long-distance cell-to-cell communication and autophagosome transfer in squamous cell carcinoma via tunneling nanotubes. Oncotarget 2017, 8, 20939–20960. [Google Scholar] [CrossRef] [Green Version]
- Spray, D.C.; Hanstein, R.; Lopez-Quintero, S.V.; Stout, R.F., Jr.; Suadicani, S.O.; Thi, M.M. Gap junctions and Bystander Effects: Good Samaritans and executioners. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2013, 2, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [Green Version]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Rhett, J.M.; Calder, B.W.; Fann, S.A.; Bainbridge, H.; Gourdie, R.G.; Yost, M.J. Mechanism of action of the anti-inflammatory connexin43 mimetic peptide JM2. Am. J. Physiol. Cell Physiol. 2017, 313, C314–C326. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, M.; Piperigkou, Z.; Riti, E.; Masola, V.; Onisto, M.; Karamanos, N.K. Long filopodia and tunneling nanotubes define new phenotypes of breast cancer cells in 3D cultures. Matrix Biol. Plus 2020, 6–7, 100026. [Google Scholar] [CrossRef]
- Pulze, L.; Congiu, T.; Brevini, T.A.L.; Grimaldi, A.; Tettamanti, G.; D’Antona, P.; Baranzini, N.; Acquati, F.; Ferraro, F.; de Eguileor, M. MCF7 Spheroid Development: New Insight about Spatio/Temporal Arrangements of TNTs, Amyloid Fibrils, Cell Connections, and Cellular Bridges. Int. J. Mol. Sci. 2020, 21, 5400. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayanithy, V.; Babatunde, V.; Dickson, E.L.; Wong, P.; Oh, S.; Ke, X.; Barlas, A.; Fujisawa, S.; Romin, Y.; Moreira, A.L.; et al. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 2014, 323, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, F.; Jean-Jacques, B.; Roberge, H.; Bénard, M.; Galas, L.; Schapman, D.; Elie, N.; Goux, D.; Keller, M.; Maille, E.; et al. A role for RASSF1A in tunneling nanotube formation between cells through GEFH1/Rab11 pathway control. Cell Commun. Signal. 2018, 16, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilleron, J.; Carette, D.; Fiorini, C.; Dompierre, J.; Macia, E.; Denizot, J.-P.; Segretain, D.; Pointis, G. The large GTPase dynamin2: A new player in connexin 43 gap junction endocytosis, recycling and degradation. Int. J. Biochem. Cell Biol. 2011, 43, 1208–1217. [Google Scholar] [CrossRef]
- Zhu, S.; Bhat, S.; Syan, S.; Kuchitsu, Y.; Fukuda, M.; Zurzolo, C. Rab11a-Rab8a cascade regulates the formation of tunneling nanotubes through vesicle recycling. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Guo, Y.; Yang, W.; Chen, L.; Ren, D.; Wu, C.; He, B.; Zheng, P.; Tong, W. Phosphorylation of connexin 43 induced by traumatic brain injury promotes exosome release. J. Neurophysiol. 2018, 119, 305–311. [Google Scholar] [CrossRef]
- Batista-Almeida, D.; Ribeiro-Rodrigues, T.; Martins-Marques, T.; Cortes, L.; Antunes, M.J.; Antunes, P.E.; Gonçalves, L.; Brou, C.; Aasen, T.; Zurzolo, C.; et al. Ischaemia impacts TNT-mediated communication between cardiac cells. Curr. Res. Cell Biol. 2020, 1, 100001. [Google Scholar] [CrossRef]
- Aasen, T.; Izpisúa Belmonte, J.C. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat. Protoc. 2010, 5, 371–382. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tishchenko, A.; Azorín, D.D.; Vidal-Brime, L.; Muñoz, M.J.; Arenas, P.J.; Pearce, C.; Girao, H.; Ramón y Cajal, S.; Aasen, T. Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells. Cancers 2020, 12, 2798. https://doi.org/10.3390/cancers12102798
Tishchenko A, Azorín DD, Vidal-Brime L, Muñoz MJ, Arenas PJ, Pearce C, Girao H, Ramón y Cajal S, Aasen T. Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells. Cancers. 2020; 12(10):2798. https://doi.org/10.3390/cancers12102798
Chicago/Turabian StyleTishchenko, Alexander, Daniel D. Azorín, Laia Vidal-Brime, María José Muñoz, Pol Jiménez Arenas, Christopher Pearce, Henrique Girao, Santiago Ramón y Cajal, and Trond Aasen. 2020. "Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells" Cancers 12, no. 10: 2798. https://doi.org/10.3390/cancers12102798
APA StyleTishchenko, A., Azorín, D. D., Vidal-Brime, L., Muñoz, M. J., Arenas, P. J., Pearce, C., Girao, H., Ramón y Cajal, S., & Aasen, T. (2020). Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells. Cancers, 12(10), 2798. https://doi.org/10.3390/cancers12102798