Oncogenic Tyrosine Phosphatases: Novel Therapeutic Targets for Melanoma Treatment

 ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract
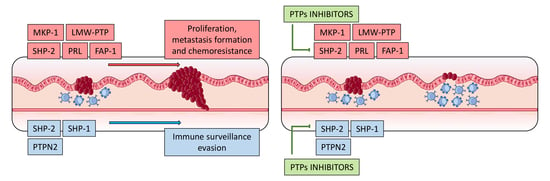
1. Introduction
2. Oncogenic Protein Tyrosine Phosphatases in Melanoma
2.1. Cell Division Cycle 25 Proteins (CDC25s)
2.2. Low-Molecular-Weight Protein Tyrosine Phosphatase (LMW-PTP)
2.3. FAS-Associated Phosphatase 1 (FAP-1)
2.4. Mitogen-Activated Protein Kinase Phosphatase-1 (MKP1)
2.5. Phosphatase of Regenerating Liver (PRL)
2.6. Src Homology Region 2 Domain-Containing Phosphatase-2 (SHP-2)
3. Role of PTPs in Immune Melanoma Cell Infiltrate
3.1. Src Homology Region 2 Domain-Containing Phosphatase-1 (SHP-1)
3.2. Src Homology Region 2 Domain-Containing Phosphatase-2 (SHP-2)
3.3. Tyrosine-Protein Phosphatase Non-Receptor Type 2 (PTPN2)
4. Discussion
5. Conclusions
Funding
Conflicts of Interest
References
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L. Lo The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Östman, A. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001, 11, 258–266. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Motiwala, T.; Jacob, S.T. Role of Protein Tyrosine Phosphatases in Cancer. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 297–329. [Google Scholar] [PubMed] [Green Version]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, A. Targeting Tyrosine Kinases in Cancer: Lessons for an Effective Targeted Therapy in the Clinic. Cancers 2019, 11, 490. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Baek, M.; Kim, D.J. Protein Tyrosine Signaling and its Potential Therapeutic Implications in Carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226–4246. [Google Scholar] [CrossRef]
- Alonso, A.; Nunes-Xavier, C.E.; Bayón, Y.; Pulido, R. The extended family of protein tyrosine phosphatases. In Protein Tyrosine Phosphatases; Humana Press: New York, NY, USA, 2016; pp. 1–23. [Google Scholar]
- Julien, S.G.; Dubé, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef]
- Ventura, J.-J.; Nebreda, Á.R. Protein kinases and phosphatases as therapeutic targets in cancer. Clin. Transl. Oncol. 2006, 8, 153–160. [Google Scholar] [CrossRef]
- Zhang, Z.-Y. Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc. Chem. Res. 2017, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr. Pharm. Des. 2010, 16, 1843–1862. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.M.; Bottini, N. Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci. 2017, 38, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.J. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2010, 2, 1563–1576. [Google Scholar] [CrossRef]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Møller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [Green Version]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.F.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Müller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, H.R.; Pireddu, R.; Chen, L.; Luo, Y.; Sung, S.-S.; Szymanski, A.M.; Yip, M.L.R.; Guida, W.C.; Sebti, S.M.; Wu, J.; et al. Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J. Med. Chem. 2008, 51, 4948–4956. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.-P.; Fedorov, A.A.; Lee, S.-Y.; Guo, X.-L.; Shen, K.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.-Y. Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J. Biol. Chem. 2003, 278, 12406–12414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Frijhoff, J.; Sandin, A.; Böhmer, F.-D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohayon, S.; Refua, M.; Hendler, A.; Aharoni, A.; Brik, A. Harnessing the oxidation susceptibility of deubiquitinases for inhibition with small molecules. Angew. Chem. Int. Ed. Engl. 2015, 54, 599–603. [Google Scholar] [CrossRef]
- Gopinath, P.; Mahammed, A.; Ohayon, S.; Gross, Z.; Brik, A. Understanding and predicting the potency of ROS-based enzyme inhibitors, exemplified by naphthoquinones and ubiquitin specific protease-2. Chem. Sci. 2016, 7, 7079–7086. [Google Scholar] [CrossRef] [Green Version]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P., Jr. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef] [Green Version]
- Daouti, S.; Li, W.; Qian, H.; Huang, K.-S.; Holmgren, J.; Levin, W.; Reik, L.; McGady, D.L.; Gillespie, P.; Perrotta, A.; et al. A Selective Phosphatase of Regenerating Liver Phosphatase Inhibitor Suppresses Tumor Cell Anchorage-Independent Growth by a Novel Mechanism Involving p130Cas Cleavage. Cancer Res. 2008, 68, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Lori, G.; Paoli, P.; Caselli, A.; Cirri, P.; Marzocchini, R.; Mangoni, M.; Talamonti, C.; Livi, L.; Raugei, G. Targeting LMW-PTP to sensitize melanoma cancer cells toward chemo- and radiotherapy. Cancer Med. 2018, 7, 1933–1943. [Google Scholar] [CrossRef]
- Tang, L.; Li, G.; Tron, V.A.; Trotter, M.J.; Ho, V.C. Expression of cell cycle regulators in human cutaneous malignant melanoma. Melanoma Res. 1999, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-P.; Chiu, H.-Y.; Hsiao, T.-L.; Hsiao, C.-H.; Lin, C.-C.; Liao, Y.-H. Scalp melanoma in a woman with LEOPARD syndrome: Possible implication of PTPN11 signaling in melanoma pathogenesis. J. Am. Acad. Dermatol. 2013, 69, e186–e187. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.S.; Roberts, E.R.; Wang, X.; Marin, E.; Park, T.D.; Son, S.; Ren, Y.; Fang, B.; Yoder, S.; Kim, S.; et al. PTPN11 Plays Oncogenic Roles and Is a Therapeutic Target for BRAF Wild-Type Melanomas. Mol. Cancer Res. 2019, 17, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.-Y.; Yu, Z.-H.; Zeng, L.; Zhang, S.; Bai, Y.; Miao, J.; Chen, L.; Xie, J.; Zhang, Z.-Y. SHP2 phosphatase as a novel therapeutic target for melanoma treatment. Oncotarget 2016, 7, 73817–73829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackett, L.A.; Scolyer, R.A. A Review of Key Biological and Molecular Events Underpinning Transformation of Melanocytes to Primary and Metastatic Melanoma. Cancers 2019, 11, 2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, R.I.; Lin, J.Y. Cutaneous Melanoma-A Review in Detection, Staging, and Management. Hematol. Oncol. Clin. North. Am. 2019, 33, 25–38. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, C.C.; Wu, X.-C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of noncutaneous melanomas in the U.S. Cancer 2005, 103, 1000–1007. [Google Scholar] [CrossRef]
- Wilkins, D.K.; Nathan, P.D. Therapeutic opportunities in noncutaneous melanoma. Ther. Adv. Med. Oncol. 2009, 1, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Rajkumar, S.; Watson, I.R. Molecular characterisation of cutaneous melanoma: Creating a framework for targeted and immune therapies. Br. J. Cancer 2016, 115, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Flaherty, K.T. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat. Rev. Clin. Oncol. 2011, 8, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, S.; Kallionpää, R.A.; Peltonen, J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp. Dermatol. 2017, 26, 645–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappetta, C.; Proietti, I.; Soccodato, V.; Puggioni, C.; Zaralli, R.; Pacini, L.; Porta, N.; Skroza, N.; Petrozza, V.; Potenza, C.; et al. BRAF and NRAS mutations are heterogeneous and not mutually exclusive in nodular melanoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2015, 23, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.; Scotto, D.; Josephs, D.H.; Mele, S.; Crescioli, S.; Bax, H.J.; Pellizzari, G.; Wynne, M.D.; Nakamura, M.; Hoffmann, R.M.; et al. BRAF inhibitors: Resistance and the promise of combination treatments for melanoma. Oncotarget 2017, 8, 78174–78192. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; Jorgenson, E.; Shen, L.; Xu, M.; North, J.P.; Shain, A.H.; Reuss, D.; Wu, H.; Robinson, W.A.; Olshen, A.; et al. Targeted Genomic Profiling of Acral Melanoma. J. Natl. Cancer Inst. 2019, 111, 1068–1077. [Google Scholar] [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat. Commun. 2018, 9, 116. [Google Scholar] [CrossRef]
- Prahallad, A.; Heynen, G.J.; Germano, G.; Willems, S.M.; Evers, B.; Vecchione, L.; Gambino, V.; Lieftink, C.; Beijersbergen, R.L.; Di Nicolantonio, F.; et al. PTPN11 is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell Rep. 2015, 12, 1978–1985. [Google Scholar] [CrossRef] [Green Version]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Damsky, W.E.; Theodosakis, N.; Bosenberg, M. Melanoma metastasis: New concepts and evolving paradigms. Oncogene 2014, 33, 2413–2422. [Google Scholar] [CrossRef] [Green Version]
- Mouawad, R.; Sebert, M.; Michels, J.; Bloch, J.; Spano, J.-P.; Khayat, D. Treatment for metastatic malignant melanoma: Old drugs and new strategies. Crit. Rev. Oncol. Hematol. 2010, 74, 27–39. [Google Scholar] [CrossRef]
- Jiang, Z.-X.; Zhang, Z.-Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Cerchia, C.; Di Giovanni, C.; Granato, G.; Albano, F.; Romano, S.; De Vendittis, E.; Ruocco, M.R.; Lavecchia, A. Ligand-based chemoinformatic discovery of a novel small molecule inhibitor targeting CDC25 dual specificity phosphatases and displaying in vitro efficacy against melanoma cells. Oncotarget 2015, 6, 40202–40222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson-Rosenthal, C.; Millar, J.B.A. Cdc25: Mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006, 16, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.-Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z.; et al. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Cangi, M.G.; Cukor, B.; Soung, P.; Signoretti, S.; Moreira, G.; Ranashinge, M.; Cady, B.; Pagano, M.; Loda, M. Role of the Cdc25A phosphatase in human breast cancer. J. Clin. Investig. 2000, 106, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-Q.; Chua, S.S.; DeMayo, F.J.; Tsai, S.Y. Induction of mammary gland hyperplasia in transgenic mice over-expressing human Cdc25B. Oncogene 1999, 18, 4564–4576. [Google Scholar] [CrossRef] [Green Version]
- Kristjánsdóttir, K.; Rudolph, J. Cdc25 Phosphatases and Cancer. Chem. Biol. 2004, 11, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Albert, H.; Santos, S.; Battaglia, E.; Brito, M.; Monteiro, C.; Bagrel, D. Differential expression of CDC25 phosphatases splice variants in human breast cancer cells. Clin. Chem. Lab. Med. 2011, 49. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Hennigan, R.F.; Myer, D.L.; Stambrook, P.J. Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation. Oncogene 2004, 23, 2658–2663. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000164045-CDC25A/pathology/melanoma (accessed on 27 August 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000101224-CDC25B/pathology/melanoma (accessed on 27 August 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000158402-CDC25C/pathology/melanoma (accessed on 27 August 2020).
- Fauman, E.B.; Cogswell, J.P.; Lovejoy, B.; Rocque, W.J.; Holmes, W.; Montana, V.G.; Piwnica-Worms, H.; Rink, M.J.; Saper, M.A. Crystal Structure of the Catalytic Domain of the Human Cell Cycle Control Phosphatase, Cdc25A. Cell 1998, 93, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, R.A.; Yem, A.W.; Wolfe, C.L.; Deibel, M.R.; Chidester, C.G.; Watenpaugh, K.D. Crystal structure of the catalytic subunit of Cdc25B required for G 2 /M phase transition of the cell cycle 1 1Edited by I. A. Wilson. J. Mol. Biol. 1999, 293, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Hung, F.-M.; Chen, Y.-L.; Huang, A.-C.; Hsiao, Y.-P.; Yang, J.-S.; Chung, M.-T.; Chueh, F.-S.; Lu, H.-F.; Chung, J.-G. Triptolide induces S phase arrest via the inhibition of cyclin E and CDC25A and triggers apoptosis via caspase- and mitochondrial-dependent signaling pathways in A375.S2 human melanoma cells. Oncol. Rep. 2013, 29, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, Y.-P.; Tsai, C.-H.; Wu, P.-P.; Hsu, S.-C.; Liu, H.-C.; Huang, Y.-P.; Yang, J.-H.; Chung, J.-G. Cantharidin induces G2/M phase arrest by inhibition of Cdc25c and Cyclin A and triggers apoptosis through reactive oxygen species and the mitochondria-dependent pathways of A375.S2 human melanoma cells. Int. J. Oncol. 2014, 45, 2393–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.; Lefterov, I.M.; Wang, M.; Lazo, J.S.; Scott, C.N.; Wilcox, C.S.; Carr, B.I. Binding and Inhibition of Cdc25 Phosphatases by Vitamin K Analogues †. Biochemistry 2003, 42, 10490–10497. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Amoscato, A.A.; Bier, M.E.; Lazo, J.S. Dual G 1 and G 2 Phase Inhibition by a Novel, Selective Cdc25 Inhibitor 7-Chloro-6-(2-morpholin-4-ylethylamino)- quinoline-5,8-dione. J. Biol. Chem. 2002, 277, 46877–46885. [Google Scholar] [CrossRef] [Green Version]
- Brisson, M.; Nguyen, T.; Wipf, P.; Joo, B.; Day, B.W.; Skoko, J.S.; Schreiber, E.M.; Foster, C.; Bansal, P.; Lazo, J.S. Redox Regulation of Cdc25B by Cell-Active Quinolinediones. Mol. Pharmacol. 2005, 68, 1810–1820. [Google Scholar] [CrossRef]
- Zhou, Y.; Feng, X.; Wang, L.; Du, J.; Zhou, Y.; Yu, H.; Zang, Y.; Li, J.; Li, J. LGH00031, a novel ortho-quinonoid inhibitor of cell division cycle 25B, inhibits human cancer cells via ROS generation. Acta Pharmacol. Sin. 2009, 30, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Cerchia, C.; Nasso, R.; Mori, M.; Villa, S.; Gelain, A.; Capasso, A.; Aliotta, F.; Simonetti, M.; Rullo, R.; Masullo, M.; et al. Discovery of Novel Naphthylphenylketone and Naphthylphenylamine Derivatives as Cell Division Cycle 25B (CDC25B) Phosphatase Inhibitors: Design, Synthesis, Inhibition Mechanism, and in Vitro Efficacy against Melanoma Cell Lines. J. Med. Chem. 2019, 62, 7089–7110. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Caselli, A.; Paoli, P.; Santi, A.; Mugnaioni, C.; Toti, A.; Camici, G.; Cirri, P. Low molecular weight protein tyrosine phosphatase: Multifaceted functions of an evolutionarily conserved enzyme. Biochim. Biophys. Acta 2016, 1864, 1339–1355. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P.; Raugei, G.; Manao, G.; Taddei, L.; Ramponi, G. Low M(r) phosphotyrosine protein phosphatase interacts with the PDGF receptor directly via its catalytic site. Biochem. Biophys. Res. Commun. 1996, 219, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P.; Marra, F.; Raugei, G.; Camici, G.; Manao, G.; Ramponi, G. LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling. Biochem. Biophys. Res. Commun. 1997, 238, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Kikawa, K.D.; Vidale, D.R.; Van Etten, R.L.; Kinch, M.S. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J. Biol. Chem. 2002, 277, 39274–39279. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, P.; Taddei, M.L.; Schiavone, N.; Papucci, L.; Giannoni, E.; Fiaschi, T.; Capaccioli, S.; Raugei, G.; Ramponi, G. LMW-PTP is a positive regulator of tumor onset and growth. Oncogene 2004, 23, 3905–3914. [Google Scholar] [CrossRef] [Green Version]
- Zambuzzi, W.F.; Granjeiro, J.M.; Parikh, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Ferreira, C.V. Modulation of Src activity by low molecular weight protein tyrosine phosphatase during osteoblast differentiation. Cell. Physiol. Biochem. 2008, 22, 497–506. [Google Scholar] [CrossRef]
- Rigacci, S.; Rovida, E.; Dello Sbarba, P.; Berti, A. Low Mr phosphotyrosine protein phosphatase associates and dephosphorylates p125 focal adhesion kinase, interfering with cell motility and spreading. J. Biol. Chem. 2002, 277, 41631–41636. [Google Scholar] [CrossRef] [Green Version]
- Caselli, A.; Taddei, M.L.; Bini, C.; Paoli, P.; Camici, G.; Manao, G.; Cirri, P.; Ramponi, G. Low molecular weight protein tyrosine phosphatase and caveolin-1: Interaction and isoenzyme-dependent regulation. Biochemistry 2007, 46, 6383–6392. [Google Scholar] [CrossRef]
- Rigacci, S.; Talini, D.; Berti, A. LMW-PTP associates and dephosphorylates STAT5 interacting with its C-terminal domain. Biochem. Biophys. Res. Commun. 2003, 312, 360–366. [Google Scholar] [CrossRef]
- Taddei, M.L.; Chiarugi, P.; Cirri, P.; Buricchi, F.; Fiaschi, T.; Giannoni, E.; Talini, D.; Cozzi, G.; Formigli, L.; Raugei, G.; et al. Β-Catenin Interacts With Low-Molecular-Weight Protein Tyrosine Phosphatase Leading To Cadherin-Mediated Cell-Cell Adhesion Increase. Cancer Res. 2002, 62, 6489–6499. [Google Scholar]
- Chiarugi, P.; Cirri, P.; Taddei, L.; Giannoni, E.; Camici, G.; Manao, G.; Raugei, G.; Ramponi, G. The low M(r) protein-tyrosine phosphatase is involved in Rho-mediated cytoskeleton rearrangement after integrin and platelet-derived growth factor stimulation. J. Biol. Chem. 2000, 275, 4640–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malentacchi, F.; Marzocchini, R.; Gelmini, S.; Orlando, C.; Serio, M.; Ramponi, G.; Raugei, G. Up-regulated expression of low molecular weight protein tyrosine phosphatases in different human cancers. Biochem. Biophys. Res. Commun. 2005, 334, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Marzocchini, R.; Malentacchi, F.; Biagini, M.; Cirelli, D.; Luceri, C.; Caderni, G.; Raugei, G. The expression of low molecular weight protein tyrosine phosphatase is up-regulated in 1,2-dimethylhydrazine-induced colon tumours in rats. Int. J. Cancer 2008, 122, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.A.; Ruela-de-Sousa, R.R.; Queiroz, K.C.S.; Souza, A.C.S.; Milani, R.; Pilli, R.A.; Peppelenbosch, M.P.; den Hertog, J.; Ferreira, C.V. Knocking down low molecular weight protein tyrosine phosphatase (LMW-PTP) reverts chemoresistance through inactivation of Src and Bcr-Abl proteins. PLoS ONE 2012, 7, e44312. [Google Scholar] [CrossRef]
- Capitani, N.; Lori, G.; Paoli, P.; Patrussi, L.; Troilo, A.; Baldari, C.T.; Raugei, G.; D’Elios, M.M. LMW-PTP targeting potentiates the effects of drugs used in chronic lymphocytic leukemia therapy. Cancer Cell Int. 2019, 19, 67. [Google Scholar] [CrossRef] [Green Version]
- Alho, I.; Costa, L.; Bicho, M.; Coelho, C. Low molecular weight protein tyrosine phosphatase isoforms regulate breast cancer cells migration through a RhoA dependent mechanism. PLoS ONE 2013, 8, e76307. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000143727-ACP1/pathology/melanoma (accessed on 27 August 2020).
- Sato, T.; Irie, S.; Kitada, S.; Reed, J. FAP-1: A protein tyrosine phosphatase that associates with Fas. Science 1995, 268, 411–415. [Google Scholar] [CrossRef]
- Nakai, Y.; Irie, S.; Sato, T.-A. Identification of IκBα as a substrate of Fas-associated phosphatase-1. Eur. J. Biochem. 2000, 267, 7170–7175. [Google Scholar] [CrossRef]
- Ungefroren, H.; Voss, M.; Jansen, M.; Roeder, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Human pancreatic adenocarcinomas express Fas and Fas ligand yet are resistant to Fas-mediated apoptosis. Cancer Res. 1998, 58, 1741–1749. [Google Scholar]
- Ungefroren, H.; Kruse, M.L.; Trauzold, A.; Roeschmann, S.; Roeder, C.; Arlt, A.; Henne-Bruns, D.; Kalthoff, H. FAP-1 in pancreatic cancer cells: Functional and mechanistic studies on its inhibitory role in CD95-mediated apoptosis. J. Cell Sci. 2001, 114, 2735–2746. [Google Scholar]
- Ivanov, V.N.; Lopez Bergami, P.; Maulit, G.; Sato, T.-A.; Sassoon, D.; Ronai, Z. FAP-1 association with Fas (Apo-1) inhibits Fas expression on the cell surface. Mol. Cell. Biol. 2003, 23, 3623–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, J.; Li, H.; Cui, Y.; Wong, A.H.Y.; Langford, C.; Tao, Q. Epigenetic disruption of two proapoptotic genes MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas through hypermethylation of a common bidirectional promoter. Leukemia 2006, 20, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, S.-H.; Wu, D.-C.; Tsai, C.-Y.; Kuo, T.-J.; Yu, W.-C.; Chang, Y.-S.; Chen, C.-L.; Chang, C.-F.; Chen, D.-S.; Chen, P.-J. Genetic characterization of fas-associated phosphatase-1 as a putative tumor suppressor gene on chromosome 4q21.3 in hepatocellular carcinoma. Clin. Cancer Res. 2006, 12, 1097–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaan, O.D.; Toretsky, J.A. PTPL1: A large phosphatase with a split personality. Cancer Metastasis Rev. 2008, 27, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in tumorigenesis, tumor progression and therapy. Cancer Med. 2016, 5, 2061–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Machado-Pinilla, R.; Manguan-García, C.; Belda-Iniesta, C.; Moratilla, C.; Cejas, P.; Fresno-Vara, J.A.; de Castro-Carpeño, J.; Casado, E.; Nistal, M.; et al. MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene 2006, 25, 3335–3345. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Zhou, J.-Y.; Liu, Y.; Wu, G.S. Mitogen-Activated Protein Kinase Phosphatase-1 Is Required for Cisplatin Resistance. Cancer Res. 2006, 66, 8870–8877. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000120129-DUSP1/pathology/melanoma (accessed on 27 August 2020).
- Liao, Q.; Guo, J.; Kleeff, J.; Zimmermann, A.; Büchler, M.W.; Korc, M.; Friess, H. Down-regulation of the dual-specificity phosphatase MKP-1 suppresses tumorigenicity of pancreatic cancer cells. Gastroenterology 2003, 124, 1830–1845. [Google Scholar] [CrossRef]
- Mizuno, R.; Oya, M.; Shiomi, T.; Marumo, K.; Okada, Y.; Murai, M. Inhibition of MKP-1 expression potentiates JNK related apoptosis in renal cancer cells. J. Urol. 2004, 172, 723–727. [Google Scholar] [CrossRef]
- Kundu, S.; Fan, K.; Cao, M.; Lindner, D.J.; Tuthill, R.; Liu, L.; Gerson, S.; Borden, E.; Yi, T. Tyrosine phosphatase inhibitor-3 sensitizes melanoma and colon cancer to biotherapeutics and chemotherapeutics. Mol. Cancer Ther. 2010, 9, 2287–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000184007-PTP4A2/pathology/melanoma (accessed on 27 August 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000184489-PTP4A3/pathology/melanoma (accessed on 27 August 2020).
- Wei, M.; Korotkov, K.V.; Blackburn, J.S. Targeting phosphatases of regenerating liver (PRLs) in cancer. Pharmacol. Ther. 2018, 190, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Duciel, L.; Monraz Gomez, L.C.; Kondratova, M.; Kuperstein, I.; Saule, S. The Phosphatase PRL-3 Is Involved in Key Steps of Cancer Metastasis. J. Mol. Biol. 2019, 431, 3056–3067. [Google Scholar] [CrossRef] [PubMed]
- McParland, V.; Varsano, G.; Li, X.; Thornton, J.; Baby, J.; Aravind, A.; Meyer, C.; Pavic, K.; Rios, P.; Köhn, M. The metastasis-promoting phosphatase PRL-3 shows activity toward phosphoinositides. Biochemistry 2011, 50, 7579–7590. [Google Scholar] [CrossRef] [PubMed]
- Fiordalisi, J.J.; Dewar, B.J.; Graves, L.M.; Madigan, J.P.; Cox, A.D. Src-Mediated Phosphorylation of the Tyrosine Phosphatase PRL-3 Is Required for PRL-3 Promotion of Rho Activation, Motility and Invasion. PLoS ONE 2013, 8, e64309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maacha, S.; Anezo, O.; Foy, M.; Liot, G.; Mery, L.; Laurent, C.; Sastre-Garau, X.; Piperno-Neumann, S.; Cassoux, N.; Planque, N.; et al. Protein Tyrosine Phosphatase 4A3 (PTP4A3) Promotes Human Uveal Melanoma Aggressiveness Through Membrane Accumulation of Matrix Metalloproteinase 14 (MMP14). Investig. Opthalmology Vis. Sci. 2016, 57, 1982. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Quah, S.Y.; Dong, J.M.; Manser, E.; Tang, J.P.; Zeng, Q. PRL-3 Down-regulates PTEN Expression and Signals through PI3K to Promote Epithelial-Mesenchymal Transition. Cancer Res. 2007, 67, 2922–2926. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Bai, Y.; Lyle, L.T.; Yu, G.; Amarasinghe, O.; Nguele Meke, F.; Carlock, C.; Zhang, Z.-Y. Mechanism of PRL2 phosphatase-mediated PTEN degradation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 20538–20548. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zeng, H.; Zhang, X.; Zhao, Y.; Sha, H.; Ge, X.; Zhang, M.; Gao, X.; Xu, Q. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am. J. Pathol. 2004, 164, 2039–2054. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Li, Y.-P.; Sheng, X.; Zhang, Z.-C.; Song, R.; Dong, W.; Cao, S.-X.; Hua, Z.-C.; Xu, Q. PRL-3 siRNA inhibits the metastasis of B16-BL6 mouse melanoma cells in vitro and in vivo. Mol. Med. 2007, 13, 151–159. [Google Scholar] [CrossRef]
- Laurent, C.; Valet, F.; Planque, N.; Silveri, L.; Maacha, S.; Anezo, O.; Hupe, P.; Plancher, C.; Reyes, C.; Albaud, B.; et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011, 71, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Duciel, L.; Anezo, O.; Mandal, K.; Laurent, C.; Planque, N.; Coquelle, F.M.; Gentien, D.; Manneville, J.-B.; Saule, S. Protein tyrosine phosphatase 4A3 (PTP4A3/PRL-3) promotes the aggressiveness of human uveal melanoma through dephosphorylation of CRMP2. Sci. Rep. 2019, 9, 2990. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.K.; Dhawan, D.; Lindner, D.J.; Borden, E.C.; Farver, C.; Yi, T. Pentamidine is an inhibitor of PRL phosphatases with anticancer activity. Mol. Cancer Ther. 2002, 1, 1255–1264. [Google Scholar] [PubMed]
- Sun, J.-P.; Luo, Y.; Yu, X.; Wang, W.-Q.; Zhou, B.; Liang, F.; Zhang, Z.-Y. Phosphatase activity, trimerization, and the C-terminal polybasic region are all required for PRL1-mediated cell growth and migration. J. Biol. Chem. 2007, 282, 29043–29051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Yu, Z.H.; Liu, S.; Zhang, L.; Zhang, R.Y.; Zeng, L.F.; Zhang, S.; Zhang, Z.Y. Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res. 2016, 76, 4805–4815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez-Mascarell, P.; González-Recio, I.; Fernández-Rodríguez, C.; Oyenarte, I.; Müller, D.; Martínez-Chantar, M.; Martínez-Cruz, L. Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators. Int. J. Mol. Sci. 2019, 20, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, S.; Kostantin, E.; Hatzihristidis, T.; Zolotarov, Y.; Uetani, N.; Tremblay, M.L. Physiological and oncogenic roles of the PRL phosphatases. FEBS J. 2018, 285, 3886–3908. [Google Scholar] [CrossRef] [Green Version]
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbé, D.P.; Bégin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 2015, 34, 986–995. [Google Scholar] [CrossRef]
- Thura, M.; Al-Aidaroos, A.Q.; Gupta, A.; Chee, C.E.; Lee, S.C.; Hui, K.M.; Li, J.; Guan, Y.K.; Yong, W.P.; So, J.; et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat. Commun. 2019, 10, 2484. [Google Scholar] [CrossRef]
- Freeman, R.M.; Plutzky, J.; Neel, B.G. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: A putative homolog of Drosophila corkscrew. Proc. Natl. Acad. Sci. USA 1992, 89, 11239–11243. [Google Scholar] [CrossRef] [Green Version]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal Structure of the Tyrosine Phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef] [Green Version]
- QU, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef]
- Hu, Z.-Q.; Ma, R.; Zhang, C.; Li, J.; Li, L.; Hu, Z.-T.; Gao, Q.; Li, W.-M. Expression and clinical significance of tyrosine phosphatase SHP2 in thyroid carcinoma. Oncol. Lett. 2015, 10, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Li, F.-Q.; Zhang, Q.; Lv, K.-Z.; Yang, H.-L.; Gao, Y.; Yu, J.-R. Expression and Clinical Significance of SHP2 in Gastric Cancer. J. Int. Med. Res. 2012, 40, 2083–2089. [Google Scholar] [CrossRef]
- Leibowitz, M.S.; Srivastava, R.M.; Andrade Filho, P.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 Is Overexpressed and Inhibits pSTAT1-Mediated APM Component Expression, T-cell Attracting Chemokine Secretion, and CTL Recognition in Head and Neck Cancer Cells. Clin. Cancer Res. 2013, 19, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Fang, H.; Wang, X.; Chen, D.; Chen, Z.; Wang, S. Overexpression of SHP2 tyrosine phosphatase promotes the tumorigenesis of breast carcinoma. Oncol. Rep. 2014, 32, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Huang, S.; Li, W.; Zhao, H.; Zhang, T.; Zhang, D. Upregulation of Src homology phosphotyrosyl phosphatase 2 (Shp2) expression in oral cancer and knockdown of Shp2 expression inhibit tumor cell viability and invasion in vitro. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Xiang, D.-M.; Sun, W.; Liu, N.; Sun, H.-L.; Wen, W.; Shen, W.-F.; Wang, R.-Y.; Chen, C.; Wang, X.; et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J. Hepatol. 2015, 63, 651–660. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, S.; Huang, Y.; Song, L.; Yin, Y.; Kong, W.; Chen, X.; Ouyang, X. Expression and prognosis value of SHP2 in patients with pancreatic ductal adenocarcinoma. Tumor Biol. 2016, 37, 7853–7859. [Google Scholar] [CrossRef]
- Zhang, K.; Zhao, H.; Ji, Z.; Zhang, C.; Zhou, P.; Wang, L.; Chen, Q.; Wang, J.; Zhang, P.; Chen, Z.; et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene 2016, 35, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000179295-PTPN11/pathology/melanoma (accessed on 27 August 2020).
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.-Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.D. Human leishmaniasis: Clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin. Infect. Dis. 1997, 24, 684–703. [Google Scholar] [CrossRef] [Green Version]
- Pathak, M.K.; Yi, T. Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J. Immunol. 2001, 167, 3391–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.; Pathak, M.K.; Lindner, D.J.; Ketterer, M.E.; Farver, C.; Borden, E.C. Anticancer Activity of Sodium Stibogluconate in Synergy with IFNs. J. Immunol. 2002, 169, 5978–5985. [Google Scholar] [CrossRef] [Green Version]
- Win-Piazza, H.; Schneeberger, V.E.; Chen, L.; Pernazza, D.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Enhanced anti-melanoma efficacy of interferon alfa-2b via inhibition of Shp2. Cancer Lett. 2012, 320, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, J.; Scott, G. Plexin B1 inhibits MET through direct association and regulates Shp2 expression in melanocytes. J. Cell Sci. 2013, 126, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Reed, S.A.; Johnson, S.E. Hepatocyte growth factor (HGF) signals through SHP2 to regulate primary mouse myoblast proliferation. Exp. Cell Res. 2009, 315, 2284–2292. [Google Scholar] [CrossRef] [Green Version]
- Schaeper, U.; Gehring, N.H.; Fuchs, K.P.; Sachs, M.; Kempkes, B.; Birchmeier, W. Coupling of Gab1 to C-Met, Grb2, and Shp2 Mediates Biological Responses. J. Cell Biol. 2000, 149, 1419–1432. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019, 26, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K.S.; Rosário, M.; Birchmeier, C.; Birchmeier, W. The Tyrosine Phosphatase Shp2 in Development and Cancer. Adv. Cancer Res. 2010, 106, 53–89. [Google Scholar] [PubMed]
- Zeng, L.-F.; Zhang, R.-Y.; Yu, Z.-H.; Li, S.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Mali, R.S.; Li, X.; Chan, R.J.; et al. Therapeutic Potential of Targeting the Oncogenic SHP2 Phosphatase. J. Med. Chem. 2014, 57, 6594–6609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaMarche, M.J.; Acker, M.G.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.; Chen, Y.-N.P.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, F.; Shields, B.; Makhoul, I.; Avaritt, N.; Wong, H.K.; Hutchins, L.F.; Shalin, S.; Tackett, A.J. Immune surveillance in melanoma: From immune attack to melanoma escape and even counterattack. Cancer Biol. Ther. 2017, 18, 451–469. [Google Scholar] [CrossRef] [Green Version]
- Shresta, S.; Pham, C.T.; Thomas, D.A.; Graubert, T.A.; Ley, T.J. How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol. 1998, 10, 581–587. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.A.G. Immunotherapy of melanoma. EJC Suppl. 2013, 11, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Pembrolizumab: A Review in Advanced Melanoma. Drugs 2016, 76, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Atrash, S.; Makhoul, I.; Mizell, J.S.; Hutchins, L.; Mahmoud, F. Response of metastatic mucosal melanoma to immunotherapy: It can get worse before it gets better. J. Oncol. Pharm. Pract. 2017, 23, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Johnson, D.B.; Peng, C.; Sosman, J.A. Nivolumab in melanoma: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2015, 7, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef] [Green Version]
- Dempke, W.C.M.; Uciechowski, P.; Fenchel, K.; Chevassut, T. Targeting SHP-1, 2 and SHIP Pathways: A novel strategy for cancer treatment? Oncology 2018, 95, 257–269. [Google Scholar] [CrossRef]
- Yang, J.; Liu, L.; He, D.; Song, X.; Liang, X.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 2003, 278, 6516–6520. [Google Scholar] [CrossRef] [Green Version]
- Varone, A.; Spano, D.; Corda, D. Shp1 in Solid Cancers and Their Therapy. Front. Oncol. 2020, 10, 935. [Google Scholar] [CrossRef]
- Takeuchi, S.; Matsushita, M.; Zimmermann, M.; Ikezoe, T.; Komatsu, N.; Seriu, T.; Schrappe, M.; Bartram, C.R.; Koeffler, H.P. Clinical significance of aberrant DNA methylation in childhood acute lymphoblastic leukemia. Leuk. Res. 2011, 35, 1345–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yang, L.; Pan, Y.; Yang, J.; Shang, Y.; Luo, J. Methylation and decreased expression of SHP-1 are related to disease progression in chronic myelogenous leukemia. Oncol. Rep. 2014, 31, 2438–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küçük, C.; Hu, X.; Jiang, B.; Klinkebiel, D.; Geng, H.; Gong, Q.; Bouska, A.; Iqbal, J.; Gaulard, P.; McKeithan, T.W.; et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin. Cancer Res. 2015, 21, 1699–1711. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Chen, X.; Wang, Y.; Liu, H.; Song, W.; Li, L.; Wang, G.; Song, J.; Shao, Z.; Fu, R. Plasma DNA methylation of p16 and shp1 in patients with B cell non-Hodgkin lymphoma. Int. J. Clin. Oncol. 2017, 22, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yaming, W.; Sun, X.; Ji, N.; Sun, S.; Yajie, W.; Liu, F.; Cui, Q.; Chen, W.; Liu, Y. Promoter methylation attenuates SHP1 expression and function in patients with primary central nervous system lymphoma. Oncol. Rep. 2017, 37, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, M.K.; Park, J.-J.; Yoo, H.S.; Lee, B.J.; Chun, H.J.; Lee, S.W.; Bak, Y.-T. Epigenetic regulation and anti-tumorigenic effects of SH2-containing protein tyrosine phosphatase 1 (SHP1) in human gastric cancer cells. Tumor Biol. 2016, 37, 4603–4612. [Google Scholar] [CrossRef]
- Sheng, Y.; Wang, H.; Liu, D.; Zhang, C.; Deng, Y.; Yang, F.; Zhang, T.; Zhang, C. Methylation of tumor suppressor gene CDH13 and SHP1 promoters and their epigenetic regulation by the UHRF1/PRMT5 complex in endometrial carcinoma. Gynecol. Oncol. 2016, 140, 145–151. [Google Scholar] [CrossRef]
- Tassidis, H.; Brokken, L.J.; Jirström, K.; Ehrnström, R.; Pontén, F.; Ulmert, D.; Bjartell, A.; Härkönen, P.; Wingren, A.G. Immunohistochemical detection of tyrosine phosphatase SHP-1 predicts outcome after radical prostatectomy for localized prostate cancer. Int. J. Cancer 2010, 126, 2296–2307. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, D.; Zhao, H.; Wu, X.; Zhao, W.; Wang, Y.; Xia, B.; Da, W. Hypermethylation of SHP-1 promoter in patient with high-risk myelodysplastic syndrome and it predicts poor prognosis. Med. Oncol. 2012, 29, 2359–2363. [Google Scholar] [CrossRef]
- Chanida, V.; Poonchavist, C.; Virote, S.; Apiwat, M. The role of SHP-1 promoter 2 hypermethylation detection of lymph node micrometastasis in resectable stage I non-small cell lung cancer as a prognostic marker of disease recurrence. Int. J. Clin. Oncol. 2014, 19, 586–592. [Google Scholar] [CrossRef]
- Tibaldi, E.; Zonta, F.; Bordin, L.; Magrin, E.; Gringeri, E.; Cillo, U.; Idotta, G.; Pagano, M.A.; Brunati, A.M. The tyrosine phosphatase SHP-1 inhibits proliferation of activated hepatic stellate cells by impairing PDGF receptor signaling. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Delesque, N.; Lopez, F.; Saint-Laurent, N.; Estève, J.-P.; Bedecs, K.; Buscail, L.; Vaysse, N.; Susini, C. sst2 Somatostatin Receptor Mediates Negative Regulation of Insulin Receptor Signaling through the Tyrosine Phosphatase SHP-1. J. Biol. Chem. 1998, 273, 7099–7106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagami, H.; Cui, T.-X.; Iwai, M.; Shiuchi, T.; Takeda-Matsubara, Y.; Wu, L.; Horiuchi, M. Tumor Necrosis Factor-α Inhibits Growth Factor–Mediated Cell Proliferation Through SHP-1 Activation in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insabato, L.; Amelio, I.; Quarto, M.; Zannetti, A.; Tolino, F.; de Mauro, G.; Cerchia, L.; Riccio, P.; Baumhoer, D.; Condorelli, G.; et al. Elevated Expression of the Tyrosine Phosphatase SHP-1 Defines a Subset of High-Grade Breast Tumors. Oncology 2009, 77, 378–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, S.C.; Kwok, T.T.; Berkowitz, R.S.; Barrett, A.J.; Tsui, F.W.L. Overexpression of the Protein Tyrosine Phosphatase, Nonreceptor Type 6 (PTPN6), in Human Epithelial Ovarian Cancer. Gynecol. Oncol. 1995, 57, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Lawson, B.R.; Rutschmann, S.; Berger, M.; Eidenschenk, C.; Blasius, A.L.; Moresco, E.M.Y.; Sovath, S.; Cengia, L.; Shultz, L.D.; et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc. Natl. Acad. Sci. USA 2008, 105, 15028–15033. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.C.; Pao, L.I.; Blattman, J.N.; Greenberg, P.D. SHP-1 in T cells limits the production of CD8 effector cells without impacting the formation of long-lived central memory cells. J. Immunol. 2010, 185, 3256–3267. [Google Scholar] [CrossRef] [Green Version]
- Kilgore, N.E.; Carter, J.D.; Lorenz, U.; Evavold, B.D. Cutting edge: Dependence of TCR antagonism on Src homology 2 domain-containing protein tyrosine phosphatase activity. J. Immunol. 2003, 170, 4891–4895. [Google Scholar] [CrossRef] [Green Version]
- Watson, H.A.; Dolton, G.; Ohme, J.; Ladell, K.; Vigar, M.; Wehenkel, S.; Hindley, J.; Mohammed, R.N.; Miners, K.; Luckwell, R.A.; et al. Purity of transferred CD8(+) T cells is crucial for safety and efficacy of combinatorial tumor immunotherapy in the absence of SHP-1. Immunol. Cell Biol. 2016, 94, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Sathish, J.G.; Dolton, G.; Leroy, F.G.; Matthews, R.J. Loss of Src homology region 2 domain-containing protein tyrosine phosphatase-1 increases CD8+ T cell-APC conjugate formation and is associated with enhanced in vivo CTL function. J. Immunol. 2007, 178, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Fowler, C.; Casamina, C.C.; Georgopolos, C.M.; McAfee, M.S.; Schmitt, T.M.; Tan, X.; Kim, T.-D.; Choi, I.; Blattman, J.N.; et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J. Immunol. 2012, 189, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.-H.; Zhang, Z.-Y. Regulatory Mechanisms and Novel Therapeutic Targeting Strategies for Protein Tyrosine Phosphatases. Chem. Rev. 2018, 118, 1069–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naing, A.; Reuben, J.M.; Camacho, L.H.; Gao, H.; Lee, B.-N.; Cohen, E.N.; Verschraegen, C.; Stephen, S.; Aaron, J.; Hong, D.; et al. Phase I Dose Escalation Study of Sodium Stibogluconate (SSG), a Protein Tyrosine Phosphatase Inhibitor, Combined with Interferon Alpha for Patients with Solid Tumors. J. Cancer 2011, 2, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-F.; Fouquet, S.; Chapon, M.; Salmon, H.; Regnier, F.; Labroquère, K.; Badoual, C.; Damotte, D.; Validire, P.; Maubec, E.; et al. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. PLoS ONE 2011, 6, e17621. [Google Scholar] [CrossRef]
- Kundu, S.; Fan, K.; Cao, M.; Lindner, D.J.; Zhao, Z.J.; Borden, E.; Yi, T. Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J. Immunol. 2010, 184, 6529–6536. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.R.; Song, W.; Lapteva, N.; Seethammagari, M.; Slawin, K.M.; Spencer, D.M.; Levitt, J.M. The Phosphatase Src Homology Region 2 Domain-Containing Phosphatase-1 Is an Intrinsic Central Regulator of Dendritic Cell Function. J. Immunol. 2011, 186, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Snook, J.P.; Soedel, A.J.; Ekiz, H.A.; O’Connell, R.M.; Williams, M.A. Inhibition of SHP-1 Expands the Repertoire of Antitumor T Cells Available to Respond to Immune Checkpoint Blockade. Cancer Immunol. Res. 2020, 8, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef] [Green Version]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 pathway: Signaling, cancer, and beyond. In Regulation of Cancer Immune Checkpoints; Springer: Singapore, 2020; pp. 33–59. [Google Scholar]
- Wu, Y.; Deng, W.; McGinley, E.C.; Klinke, D.J. Melanoma exosomes deliver a complex biological payload that upregulates PTPN11 to suppress T lymphocyte function. Pigment. Cell Melanoma Res. 2017, 30, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Guo, Y.; Zhang, H.; Zhang, X.; Cheng, H.; Cao, Q.; Ke, Y. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-γ production in tumor microenvironment. Oncogene 2018, 37, 5088–5100. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, A.; Kumar, S.; Nandi, D.; Kulkarni, A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv. Mater. 2019, 31, e1904364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Guo, W.; Yang, Y.; Liu, W.; Guo, L.; Gu, Y.; Shu, Y.; Wang, L.; Wu, X.; Hua, Z.; et al. Loss of SHP-2 activity in CD4+ T cells promotes melanoma progression and metastasis. Sci. Rep. 2013, 3, 2845. [Google Scholar] [CrossRef] [PubMed]
- Dubé, N.; Tremblay, M.L. Involvement of the small protein tyrosine phosphatases TC-PTP and PTP1B in signal transduction and diseases: From diabetes, obesity to cell cycle, and cancer. Biochim. Biophys. Acta Proteins Proteom. 2005, 1754, 108–117. [Google Scholar] [CrossRef]
- Doody, K.M.; Bourdeau, A.; Tremblay, M.L. T-cell protein tyrosine phosphatase is a key regulator in immune cell signaling: Lessons from the knockout mouse model and implications in human disease. Immunol. Rev. 2009, 228, 325–341. [Google Scholar] [CrossRef]
- Heinonen, K.M.; Nestel, F.P.; Newell, E.W.; Charette, G.; Seemayer, T.A.; Tremblay, M.L.; Lapp, W.S. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 2004, 103, 3457–3464. [Google Scholar] [CrossRef]
- Bourdeau, A.; Trop, S.; Doody, K.M.; Dumont, D.J.; Tremblayef, M.L. Inhibition of T Cell Protein Tyrosine Phosphatase Enhances Interleukin-18-Dependent Hematopoietic Stem Cell Expansion. Stem Cells 2013, 31, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Hauser, C.; Kahn, B.B.; Haj, F.G.; Neel, B.G.; Tonks, N.K.; Tiganis, T. Coordinated Regulation of Insulin Signaling by the Protein Tyrosine Phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 2005, 25, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Fukushima, A.; Zhang, X.; Galic, S.; Briggs, D.; Enriori, P.J.; Simonds, S.; Wiede, F.; Reichenbach, A.; Hauser, C.; et al. Elevated Hypothalamic TCPTP in Obesity Contributes to Cellular Leptin Resistance. Cell Metab. 2011, 14, 684–699. [Google Scholar] [CrossRef] [Green Version]
- Morales, L.D.; Archbold, A.K.; Olivarez, S.; Slaga, T.J.; DiGiovanni, J.; Kim, D.J. The role of T-cell protein tyrosine phosphatase in epithelial carcinogenesis. Mol. Carcinog. 2019, 58, 1640–1647. [Google Scholar] [CrossRef]
- Nishiyama-Fujita, Y.; Shimizu, T.; Sagawa, M.; Uchida, H.; Kizaki, M. The role of TC-PTP (PTPN2) in modulating sensitivity to imatinib and interferon-α in CML cell line, KT-1 cells. Leuk. Res. 2013, 37, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Veenstra, C.; Emin, S.; Dutta, C.; Pérez-Tenorio, G.; Nordenskjöld, B.; Fornander, T.; Stål, O. Loss of protein tyrosine phosphatase, non-receptor type 2 is associated with activation of AKT and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 2015, 153, 31–40. [Google Scholar] [CrossRef]
- Karlsson, E.; Veenstra, C.; Gårsjö, J.; Nordenskjöld, B.; Fornander, T.; Stål, O. PTPN2 deficiency along with activation of nuclear Akt predict endocrine resistance in breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.M.; Polsky, A.; Refaeli, Y. TC-PTP is required for the maintenance of MYC-driven B-cell lymphomas. Blood 2009, 114, 5016–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanquart, C.; Karouri, S.-E.; Issad, T. Protein tyrosine phosphatase-1B and T-cell protein tyrosine phosphatase regulate IGF-2-induced MCF-7 cell migration. Biochem. Biophys. Res. Commun. 2010, 392, 83–88. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Miller, B.C.; Yates, K.B.; Gillis, J.E.; Sen, D.R.; Gaudiano, E.F.; Al Abosy, R.; Freeman, G.J.; et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat. Immunol. 2019, 20, 1335–1347. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Amann, V.C.; Ramelyte, E.; Thurneysen, S.; Pitocco, R.; Bentele-Jaberg, N.; Goldinger, S.M.; Dummer, R.; Mangana, J. Developments in targeted therapy in melanoma. Eur. J. Surg. Oncol. 2017, 43, 581–593. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Phosphatases start shedding their stigma of undruggability. Nat. Rev. Drug Discov. 2018, 17, 847–849. [Google Scholar] [CrossRef]
- Hangan-Steinman, D.; Ho, W.C.; Shenoy, P.; Chan, B.M.; Morris, V.L. Differences in phosphatase modulation of alpha4beta1 and alpha5beta1 integrin-mediated adhesion and migration of B16F1 cells. Biochem. Cell Biol. 1999, 77, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Stefanini, L.; Williams, S.; Alonso, A.; Jascur, T.; Abraham, R.T.; Couture, C.; Mustelin, T. Activation of ZAP-70 through specific dephosphorylation at the inhibitory Tyr-292 by the low molecular weight phosphotyrosine phosphatase (LMPTP). J. Biol. Chem. 2002, 277, 24220–24224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Guo, W.; Wu, Y.; Yang, C.; Zhong, L.; Deng, G.; Zhu, Y.; Liu, W.; Gu, Y.; Lu, Y.; et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B 2019, 9, 304–315. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Number | Compound | Target | Disease | Status |
|---|---|---|---|---|
| NCT03191682 | PRL3-ZUMAB | PRL3 | Solid Tumors and Hematologic Malignancies | Phase I |
| NCT03114319 | TNO155 | SHP-2 | Non-Small Cell Lung Cancer; Esophageal Squamous Cell Cancer (SCC); Head/Neck SCC; Melanoma | Phase I |
| NCT00629200 | Sodium stibogluconate | SHP-1 | Malignant melanoma | Phase I completed |
| NCT00498979 | Sodium stibogluconate | SHP-1 | Malignant melanoma | Phase I completed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardella, E.; Pranzini, E.; Leo, A.; Taddei, M.L.; Paoli, P.; Raugei, G. Oncogenic Tyrosine Phosphatases: Novel Therapeutic Targets for Melanoma Treatment. Cancers 2020, 12, 2799. https://doi.org/10.3390/cancers12102799
Pardella E, Pranzini E, Leo A, Taddei ML, Paoli P, Raugei G. Oncogenic Tyrosine Phosphatases: Novel Therapeutic Targets for Melanoma Treatment. Cancers. 2020; 12(10):2799. https://doi.org/10.3390/cancers12102799
Chicago/Turabian StylePardella, Elisa, Erica Pranzini, Angela Leo, Maria Letizia Taddei, Paolo Paoli, and Giovanni Raugei. 2020. "Oncogenic Tyrosine Phosphatases: Novel Therapeutic Targets for Melanoma Treatment" Cancers 12, no. 10: 2799. https://doi.org/10.3390/cancers12102799
APA StylePardella, E., Pranzini, E., Leo, A., Taddei, M. L., Paoli, P., & Raugei, G. (2020). Oncogenic Tyrosine Phosphatases: Novel Therapeutic Targets for Melanoma Treatment. Cancers, 12(10), 2799. https://doi.org/10.3390/cancers12102799