Inhibition of Radiation and Temozolomide-Induced Glioblastoma Invadopodia Activity Using Ion Channel Drugs

 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
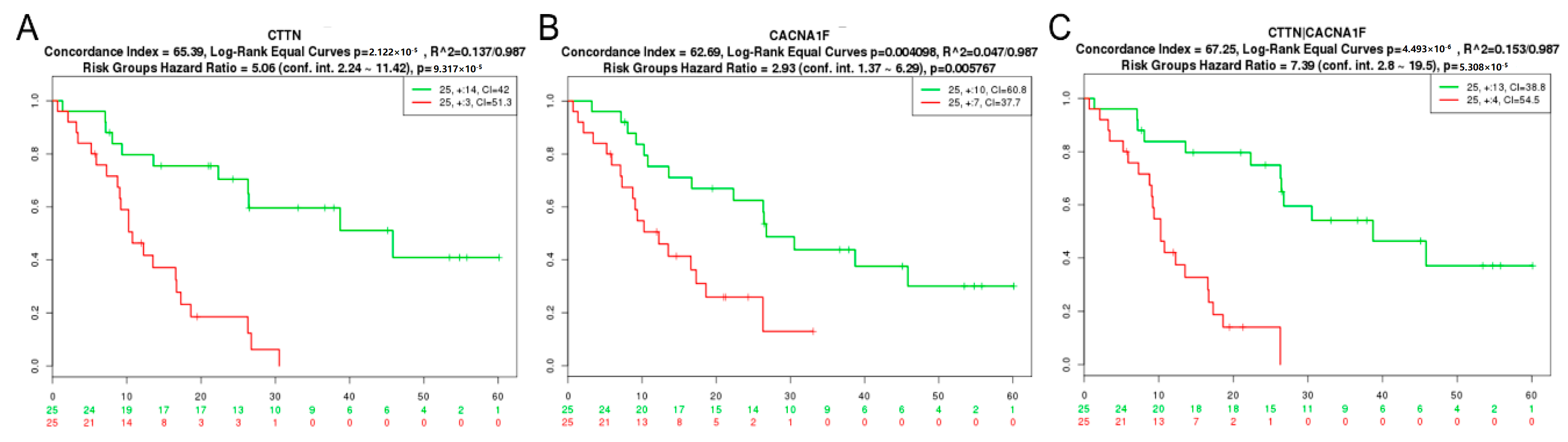
2.1. GBM Tissue Exhibits Increased Invadopodia Regulator and Ion Channel Gene Expression
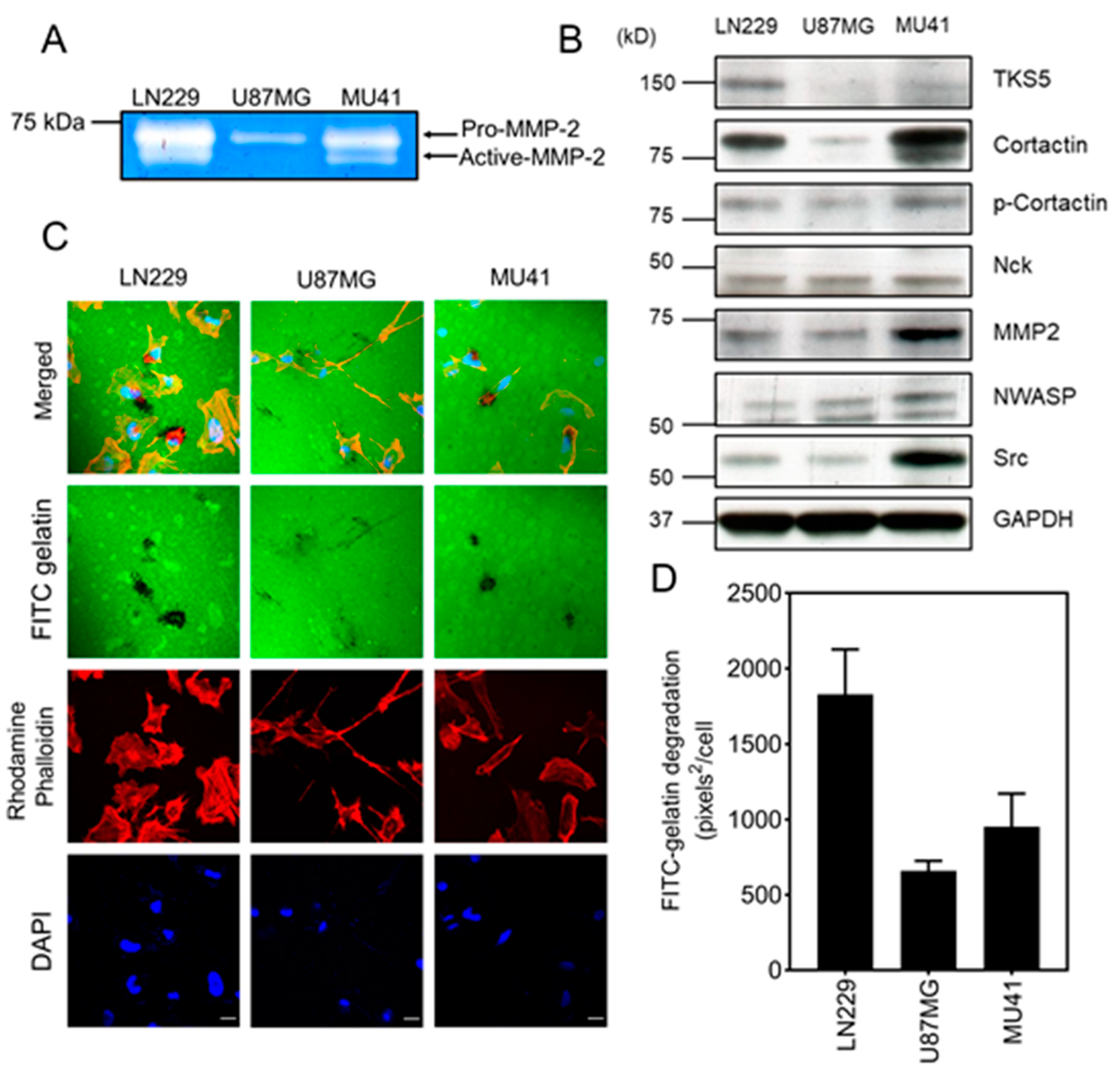
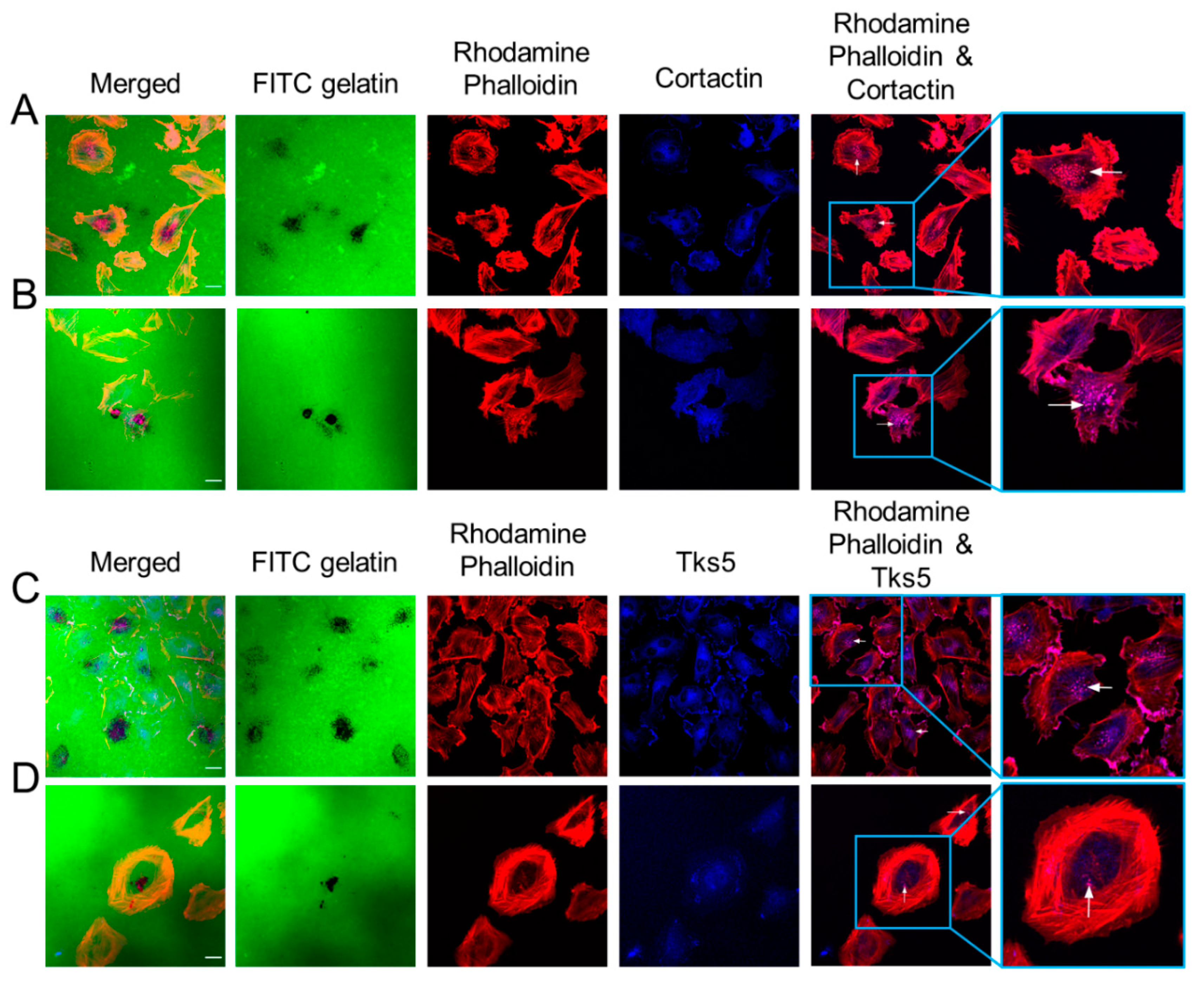
2.2. GBM Cells Form Functional Invadopodia and Express Invadopodia Regulator Proteins
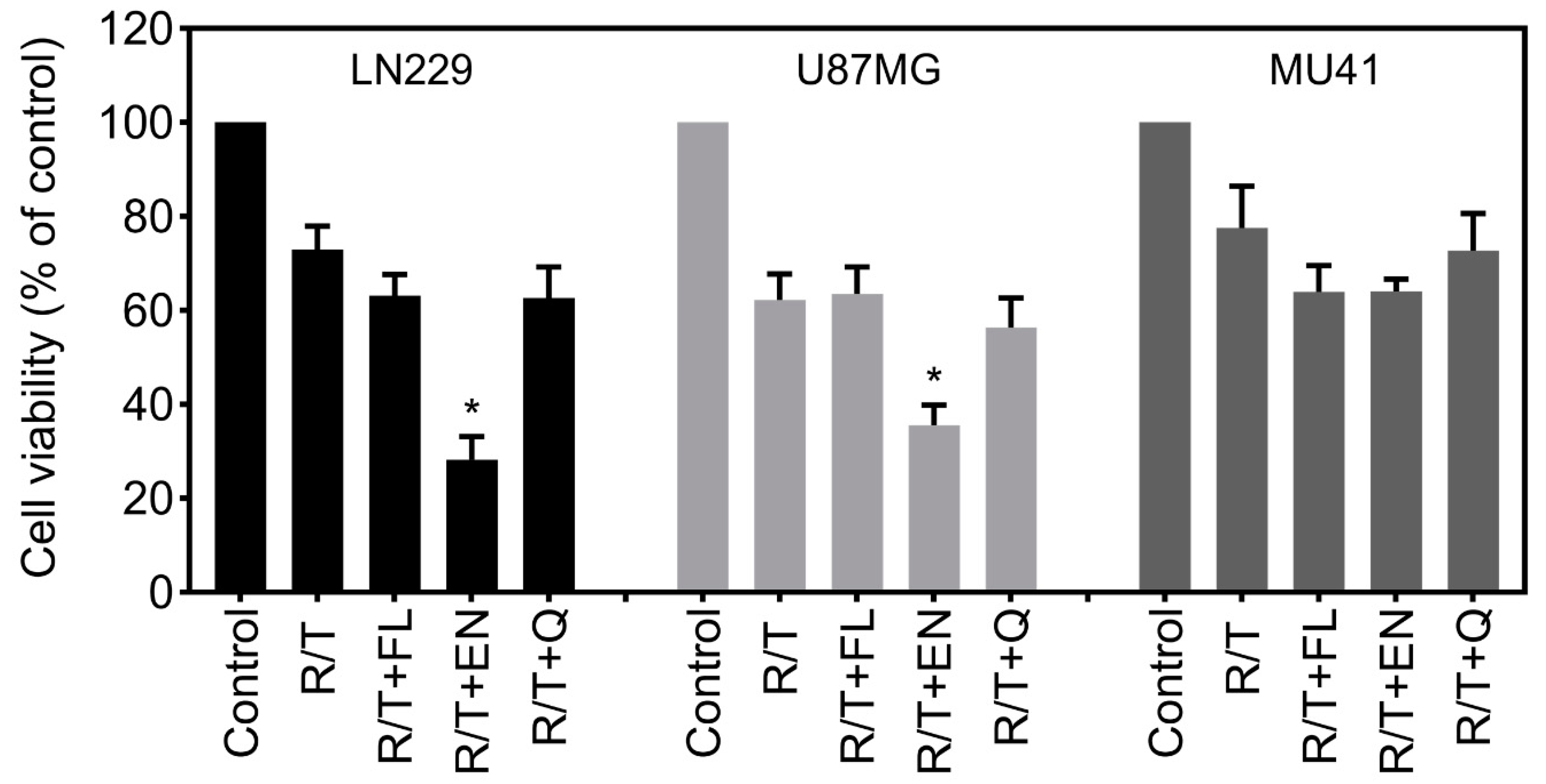
2.3. Ion Channel-Targeting Drugs Reduce GBM Cell Viability
2.4. Ion Channel Drugs Reduce MMP-2 Secretion and Invasion
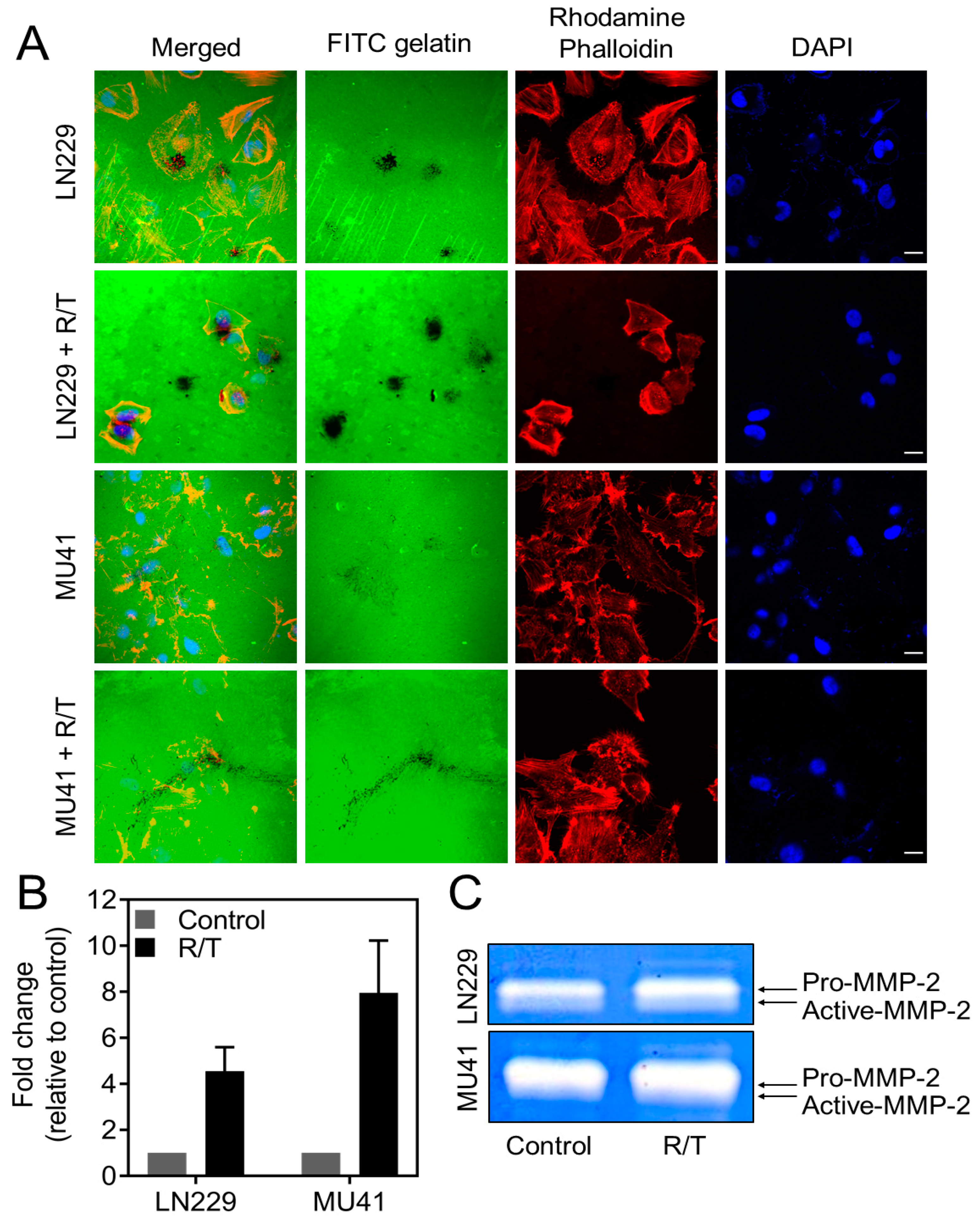
2.5. MMP-2 Secretion and Invadopodia Gelatin Degradation Is Enhanced Following Radiation and Temozolomide Treatment
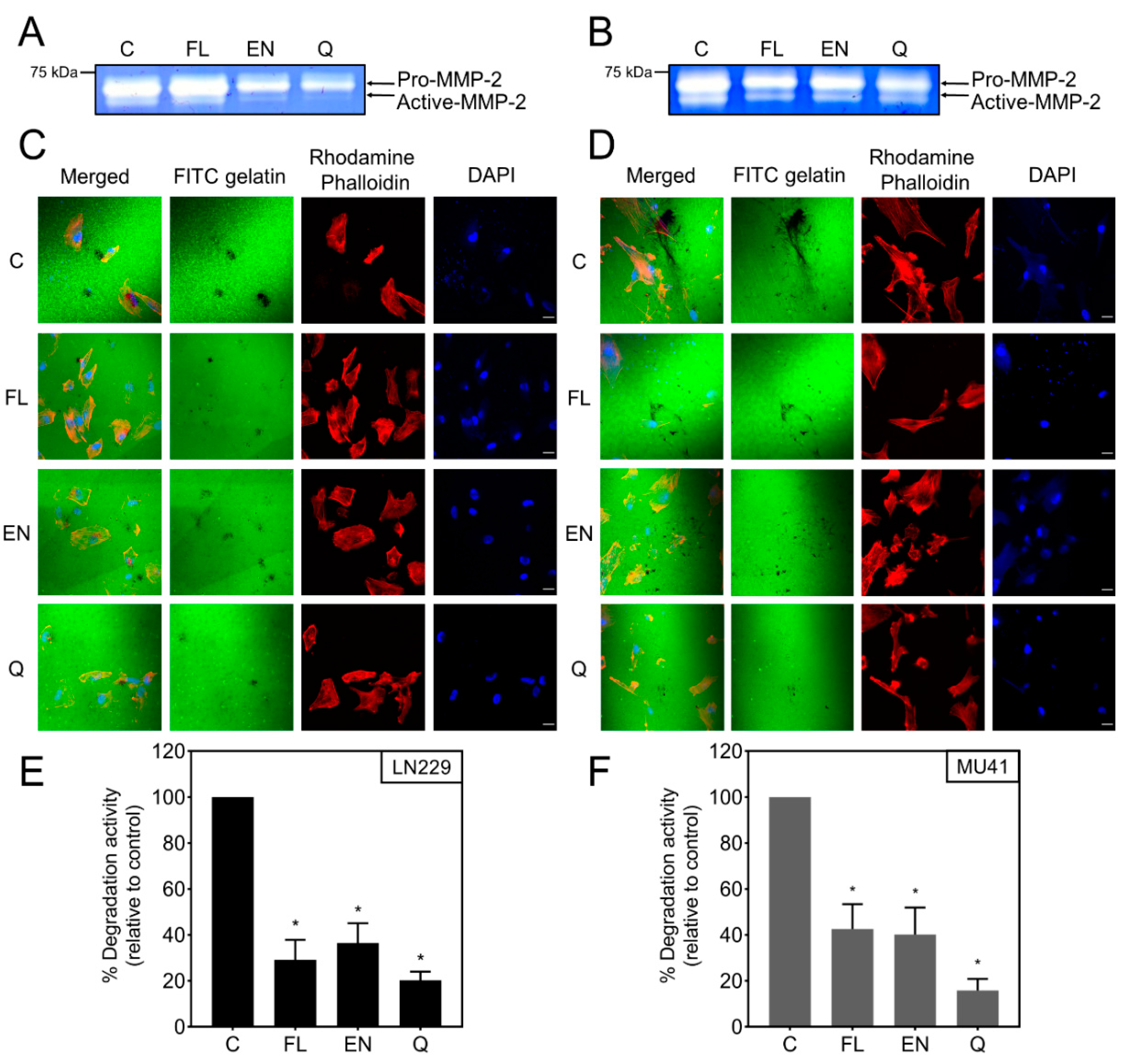
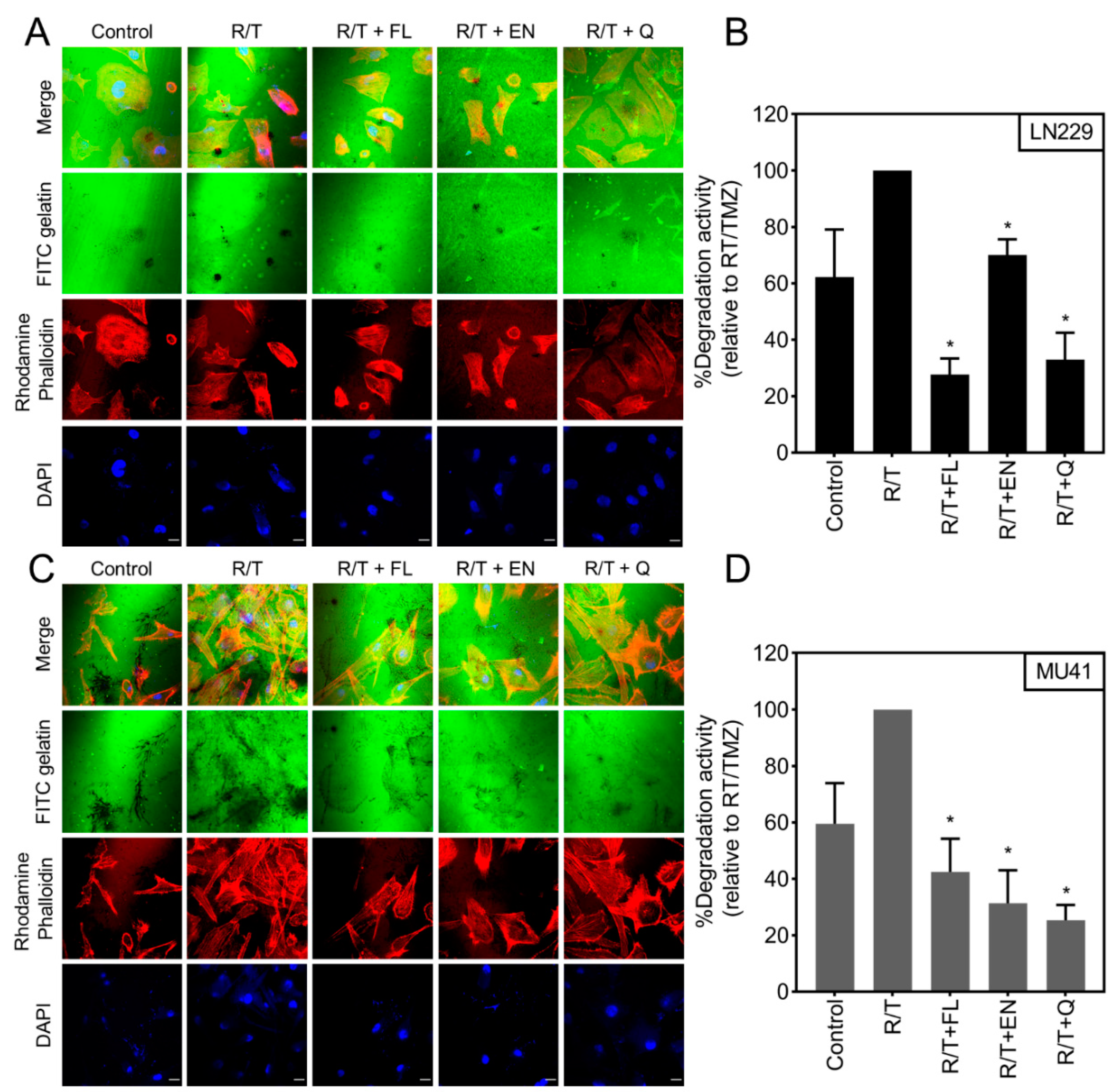
2.6. Inhibition of RT/TMZ-Induced Invadopodia Activity
3. Discussion
4. Materials and Methods
4.1. Ion Channel Drugs
4.2. Cell Lines and Cell Culture
4.3. Zymographic Analysis
4.4. Invadopodia Degradation Assay
4.5. Cell Viability
4.6. Western Blot Analysis
4.7. Oncomine Data Mining
4.8. SurvExpress
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ellor, S.V.; Pagano-Young, T.A.; Avgeropoulos, N.G. Glioblastoma: Background, standard treatment paradigms, and supportive care considerations. J. Law Med. Ethic. 2014, 42, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Zygogianni, A.; Protopapa, M.; Kougioumtzopoulou, A.; Simopoulou, F.; Nikoloudi, S.; Kouloulias, V. From imaging to biology of glioblastoma: New clinical oncology perspectives to the problem of local recurrence. Clin. Transl. Oncol. 2018, 20, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An Overview of Emerging Therapeutic Targets. Front. Oncol. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Gene Funct. Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.-W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Nakada, S.; DeMuth, T.; Tran, N.L.; Hoelzinger, D.B.; Berens, M.E. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 2007, 64, 458–478. [Google Scholar] [CrossRef] [PubMed]
- De Gooijer, M.C.; Navarro, M.G.; Bernards, R.; Wurdinger, T.; Van Tellingen, O. An Experimenter’s Guide to Glioblastoma Invasion Pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Brown, G.T.; Murray, G.I. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Artym, V.V.; Zhang, Y.; Seillier-Moiseiwitsch, F.; Yamada, K.M.; Mueller, S.C. Dynamic Interactions of Cortactin and Membrane Type 1 Matrix Metalloproteinase at Invadopodia: Defining the Stages of Invadopodia Formation and Function. Cancer Res. 2006, 66, 3034–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2013, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibony-Benyamini, H.; Gil-Henn, H. Invadopodia: The leading force. Eur. J. Cell Biol. 2012, 91, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Pixley, F.; Condeelis, J. Invadopodia and podosomes in tumor invasion. Eur. J. Cell Biol. 2006, 85, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Stylli, S.S.; Stacey, T.T.; Kaye, A.H.; Lock, P. Prognostic significance of Tks5 expression in gliomas. J. Clin. Neurosci. 2012, 19, 436–442. [Google Scholar] [CrossRef]
- Stylli, S.S.; Kaye, A.H.; Lock, P. Invadopodia: At the cutting edge of tumour invasion. J. Clin. Neurosci. 2008, 15, 725–737. [Google Scholar] [CrossRef]
- Buccione, R.; Caldieri, G.; Ayala, I. Invadopodia: Specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 2009, 28, 137–149. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.D.; Parsons, D.W.; Velculescu, V.E.; Riggins, G.J. Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Mol. Cancer 2011, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, J.; Rai, K.G.; Funk, C.C.; Arora, S.; Lee, E.; Zhu, J.; Price, N.D.; Paddison, P.J.; Ramirez, J.-M.; Rostomily, R.C. Ion channel expression patterns in glioblastoma stem cells with functional and therapeutic implications for malignancy. PLoS ONE 2017, 12, e0172884. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddapah, V.A.; Turner, K.L.; Seifert, S.; Sontheimer, H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J. Neurosci. 2013, 33, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Ecatalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, A.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef]
- Turner, K.L.; Honasoge, A.; Robert, S.M.; McFerrin, M.M.; Sontheimer, H. A proinvasive role for the Ca(2+) -activated K(+) channel KCa3.1 in malignant glioma. Glia 2014, 62, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Deliot, N.; Constantin, B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta. (BBA)-Biomembr. 2015, 1848, 2512–2522. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, J.; Jiang, D.; Zhang, N.; Qian, Z.; Liu, C.; Tao, J. Inhibition of T-type Ca2+ channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br. J. Pharmacol. 2012, 166, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Simon, O.J.; Müntefering, T.; Grauer, O.M.; Meuth, S.G. The role of ion channels in malignant brain tumors. J. Neuro-Oncol. 2015, 125, 225–235. [Google Scholar] [CrossRef]
- Stock, C.; Ludwig, F.T.; Hanley, P.J.; Schwab, A. Roles of Ion Transport in Control of Cell Motility. Compr. Physiol. 2013, 3, 59–119. [Google Scholar] [CrossRef]
- Molenaar, R.J. Ion Channels in Glioblastoma. ISRN Neurol. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P. Øyvind Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.J.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Bredel, M.; Bredel, C.; Juric, D.; Harsh, G.R.; Vogel, H.; Recht, L.D.; Sikic, B.I. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 2005, 65, 8679–8689. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Diehn, M.; Watson, N.P.; Bollen, A.W.; Aldape, K.D.; Nicholas, M.K.; Lamborn, K.R.; Berger, M.S.; Botstein, D.; Brown, P.O.; et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2005, 102, 5814–5819. [Google Scholar] [CrossRef] [Green Version]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.-F.; De Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C.; et al. Stem Cell–Related “Self-Renewal” Signature and High Epidermal Growth Factor Receptor Expression Associated with Resistance to Concomitant Chemoradiotherapy in Glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef]
- Sun, L.; Hui, A.-M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, R.; Arao, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Tanaka, R.; Sano, M.; Oide, A.; Sekijima, M.; Nishio, K. Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 2006, 25, 5994–6002. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genom. 2008, 1, 52. [Google Scholar] [CrossRef] [Green Version]
- Shai, R.; Shi, T.; Kremen, T.J.; Horvath, S.; Liau, L.M.; Cloughesy, T.F.; Mischel, P.S.; Nelson, S.F. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene 2003, 22, 4918–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutt, C.L.; Mani, D.R.; Betensky, R.A.; Tamayo, P.; Cairncross, J.G.; Ladd, C.; Pohl, U.; Hartmann, C.; McLaughlin, M.E.; Batchelor, T.; et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003, 63, 1602–1607. [Google Scholar] [PubMed]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S. Gene Expression Profiling of Gliomas Strongly Predicts Survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Saykali, B.A.; El-Sibai, M. Invadopodia, Regulation, and Assembly in Cancer Cell Invasion. Cell Commun. Adhes. 2014, 21, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Whitehead, C.A.; Paradiso, L.; Kaye, A.H.; Morokoff, A.P.; Luwor, R.B.; Stylli, S.S. Enhancement of invadopodia activity in glioma cells by sublethal doses of irradiation and temozolomide. J. Neurosurg. 2018, 129, 598–610. [Google Scholar] [CrossRef]
- Whitehead, C.A.; Nguyen, H.P.; Morokoff, A.P.; Luwor, R.B.; Paradiso, L.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S. Inhibition of Radiation and Temozolomide-Induced Invadopodia Activity in Glioma Cells Using FDA-Approved Drugs. Transl. Oncol. 2018, 11, 1406–1418. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Koltai, T. Voltage-gated sodium channel as a target for metastatic risk reduction with re-purposed drugs. F1000Research 2015, 4, 297. [Google Scholar] [CrossRef]
- Kataki, M.; T Mani Senthil Kumar, K.; Rajkumari, A. Neuropsychopharmacological profiling of flunarizine: A calcium channel blocker. Int. J. Pharmatech Res. 2010, 2, 1703–1713. [Google Scholar]
- Fischer, W.; Kittner, H.; Regenthal, R.; De Sarro, G. Anticonvulsant profile of flunarizine and relation to Na(+) channel blocking effects. Basic Clin. Pharmacol. Toxicol. 2004, 94, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.P.; Nath, A.; Haughey, N. Antifungal Agents as Neuroprotectants. US Patent 2010/0298394A1, 25 November 2010. [Google Scholar]
- Golden, E.B.; Cho, H.-Y.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Quinoline-based antimalarial drugs: A novel class of autophagy inhibitors. Neurosurg. Focus 2015, 38, E12. [Google Scholar] [CrossRef] [PubMed]
- Iamshanova, O.; Pla, A.F.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskin, V.; Szeto, A.; Ghaffari, A.; Greer, P.A.; Côté, G.P.; Elliott, B.E. Ezrin regulates focal adhesion and invadopodia dynamics by altering calpain activity to promote breast cancer cell invasion. Mol. Biol. Cell 2015, 26, 3464–3479. [Google Scholar] [CrossRef] [PubMed]
- Brisson, L.; Alfarouk, K.; Goré, J.; Roger, S. pH regulators in invadosomal functioning: Proton delivery for matrix tasting. Eur. J. Cell Biol. 2012, 91, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Wang, S.-C.; Yu, C.-F.; Hong, J.-H.; Tsai, C.-S.; Chiang, C.-S. Radiation Therapy-Induced Tumor Invasiveness Is Associated with SDF-1-Regulated Macrophage Mobilization and Vasculogenesis. PLoS ONE 2013, 8, e69182. [Google Scholar] [CrossRef] [Green Version]
- Trog, D.; Yeghiazaryan, K.; Fountoulakis, M.; Friedlein, A.; Moenkemann, H.; Haertel, N.; Schueller, H.; Breipohl, W.; Schild, H.; Leppert, D.; et al. Pro-invasive gene regulating effect of irradiation and combined temozolomide-radiation treatment on surviving human malignant glioma cells. Eur. J. Pharmacol. 2006, 542, 8–15. [Google Scholar] [CrossRef]
- Schwab, A.; Stock, C. Ion channels and transporters in tumour cell migration and invasion. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130102. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.G.; Sontheimer, H. A role for ion channels in perivascular glioma invasion. Eur. Biophys. J. 2016, 45, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Munson, J.M.; Bonner, M.Y.; Fried, L.; Hofmekler, J.; Arbiser, J.L.; Bellamkonda, R.V. Identifying new small molecule anti-invasive compounds for glioma treatment. Cell Cycle 2013, 12, 2200–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciancarelli, I.; Tozzi-Ciancarelli, M.G.; Di Massimo, C.; Marini, C.; Carolei, A. Flunarizine Effects on Oxidative Stress in Migraine Patients. Cephalalgia 2004, 24, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Bebin, M.; Bleck, T.P. New Anticonvulsant Drugs. Drugs 1994, 48, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Stylli, S.S.; Stacey, T.T.I.; Verhagen, A.M.; Xu, S.S.; Pass, I.; Courtneidge, S.A.; Lock, P.; I, S.T.T. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J. Cell Sci. 2009, 122, 2727–2740. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Trevino, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Invadopodia Marker | Number of GBM Tissue Samples | Number of Normal Tissue Samples | Total Measured Genes | Mean Fold Change (Log2) | p Value | Sample Type | Platform | Study |
|---|---|---|---|---|---|---|---|---|
| CTTN | 542 | 10 | 12,624 | 1.353 | 3 × 10−3 | mRNA | Human Genome U2A | TCGA |
| MMP2 | 27 | 4 | 14,836 | 6.426 | 5.00 × 10−4 | mRNA | ND | Bredel Brain 2 [34] |
| MMP2 | 30 | 3 | 9957 | 4.537 | 3.00 × 10−3 | mRNA | ND | Liang [35] |
| MMP2 | 80 | 4 | 19,574 | 2.92 | 2.98 × 10−4 | mRNA | Human Genome U2A | Murat [36] |
| MMP2 | 81 | 23 | 19,574 | 3.548 | 7.99 × 10−16 | mRNA | Human Genome U2A | Sun [37] |
| MMP2 | 542 | 10 | 12,624 | 4.818 | 4.06 × 10−10 | mRNA | Human Genome U2A | TCGA |
| Nck1 | 27 | 4 | 14,836 | 1.717 | 1.00 × 10−2 | mRNA | ND | Bredel Brain 2 [34] |
| Nck1 | 30 | 3 | 9957 | 1.626 | 1.90 × 10−2 | mRNA | ND | Liang [35] |
| Nck1 | 80 | 4 | 19,574 | 1.885 | 5.00 × 10−3 | mRNA | Human Genome U2A | Murat [36] |
| Nck1 | 81 | 23 | 19,574 | 1.305 | 5.41 × 10−7 | mRNA | Human Genome U2A | Sun [37] |
| Nck1 | 542 | 10 | 12,624 | 2.056 | 4.06 × 10−9 | mRNA | Human Genome U2A | TCGA |
| Nck2 | 80 | 4 | 19,574 | 1.135 | 2.00 × 10−3 | mRNA | Human Genome U2A | Murat [36] |
| NWASP | 81 | 23 | 19,574 | 1.338 | 1.10 × 10−2 | mRNA | Human Genome U2A | Sun [37] |
| Src | 80 | 4 | 19,574 | 1.035 | 4.50 × 10−2 | mRNA | Human Genome U2A | Murat [36] |
| Src | 81 | 23 | 19,574 | 1.601 | 2.00 × 10−3 | mRNA | Human Genome U2A | Sun [37] |
| Tks4 | 27 | 4 | 14,836 | 3.257 | 1.12 × 10−4 | mRNA | ND | Bredel Brain 2 [34] |
| Tks4 | 30 | 3 | 9957 | 1.492 | 1.40 × 10−2 | mRNA | ND | Liang [35] |
| Tks4 | 80 | 4 | 19,574 | 2.241 | 1.32 × 10−6 | mRNA | Human Genome U2A | Murat [36] |
| Tks4 | 81 | 23 | 19,574 | 2.194 | 2.50 × 10−4 | mRNA | Human Genome U2A | Sun [37] |
| Tks5 | 27 | 4 | 14,836 | 1.399 | 4.80 × 10−2 | mRNA | ND | Bredel Brain 2 [34] |
| Tks5 | 22 | 7 | 7689 | 1.263 | 5.00 × 10−3 | mRNA | ND | Yamanaka [38] |
| Ion Channel Gene | Number of GBM Tissue Samples | Number of Normal Tissue Samples | Total Measured Genes | Mean Fold Change (Log2) | p Value | Sample Type | Platform | Study |
|---|---|---|---|---|---|---|---|---|
| KCNH2 | 106 | 32 | 18,823 | 1.175 | 3.49 × 10−13 | DNA | RefSeq Genes | Beroukhim Brain [39] |
| KCNA5 | 107 | 32 | 18,823 | 1.054 | 2.15 × 10−4 | DNA | RefSeq Genes | Beroukhim Brain [39] |
| KCNJ10 | 107 | 33 | 18,823 | 1.041 | 2.00 × 10−3 | DNA | RefSeq Genes | Beroukhim Brain [39] |
| KCNB1 | 107 | 33 | 18,823 | 1.064 | 2.00 × 10−3 | DNA | RefSeq Genes | Beroukhim Brain [39] |
| CACNA1S | 107 | 33 | 18,823 | 1.041 | 3.20 × 10−2 | DNA | RefSeq Genes | Beroukhim Brain [39] |
| CACNA1C | 107 | 33 | 18,823 | 1.041 | 1.20 × 10−2 | DNA | RefSeq Genes | Beroukhim Brain [39] |
| KCNN4 | 21 | 3 | 14,836 | 2.219 | 1.40 × 10−2 | mRNA | ND | Bredel Brain 2 [34] |
| CACNA1D | 27 | 4 | 14,836 | 1.377 | 6.00 × 10−3 | mRNA | ND | Bredel Brain 2 [34] |
| CACNA1C | 22 | 3 | 19,574 | 8.62 | 6.00 × 10−3 | mRNA | Human Genome U2A | Lee Brain [40] |
| KCNH2 | 22 | 3 | 19,574 | 1.869 | 1.00 × 10−3 | mRNA | Human Genome U2A | Lee Brain [40] |
| KCNB1 | 22 | 3 | 19,574 | 2.325 | 2.10 × 10−2 | mRNA | Human Genome U2A | Lee Brain [40] |
| CACNA1D | 22 | 3 | 19,574 | 3.293 | 3.85 × 10−4 | mRNA | Human Genome U2A | Lee Brain [40] |
| KCNH2 | 80 | 4 | 19,574 | 1.252 | 5.00 × 10−3 | mRNA | Human Genome U2A | Murat Brain [36] |
| KCNH2 | 27 | 7 | 8603 | 1.101 | 1.20 × 10−2 | mRNA | Human Genome U95A | Shai Brain [41] |
| KCNH2 | 81 | 23 | 19,574 | 2.142 | 1.70 × 10−2 | mRNA | Human Genome U2A | Sun Brain [37] |
| KCNH2 | 542 | 10 | 12,624 | 1.094 | 1.20 × 10−2 | mRNA | Human Genome U2A | TCGA |
| KCNH2 | 582 | 37 | 18,823 | 1.320 | 4.80 × 10−169 | DNA | RefSeq Genes | TCGA 2 |
| KCNA5 | 582 | 37 | 18,823 | 1.034 | 5.75 × 10−8 | DNA | RefSeq Genes | TCGA 2 |
| KCNJ10 | 582 | 37 | 18,823 | 1.061 | 1.65 × 10−46 | DNA | RefSeq Genes | TCGA 2 |
| KCNB1 | 582 | 37 | 18,823 | 1.126 | 3.54 × 10−68 | DNA | RefSeq Genes | TCGA 2 |
| KCNN4 | 582 | 37 | 18,823 | 1.046 | 6.02 × 10−10 | DNA | RefSeq Genes | TCGA 2 |
| SCN5A | 582 | 37 | 18,823 | 1.019 | 6.64 × 10−5 | DNA | RefSeq Genes | TCGA 2 |
| SCN8A | 582 | 37 | 18,823 | 1.012 | 1.60 × 10−2 | DNA | RefSeq Genes | TCGA 2 |
| CACNA1S | 582 | 37 | 18,823 | 1.055 | 2.28 × 10−30 | DNA | RefSeq Genes | TCGA 2 |
| CACNA1C | 582 | 37 | 18,823 | 1.032 | 1.11 × 10−7 | DNA | RefSeq Genes | TCGA 2 |
| CACNA1D | 582 | 37 | 18,823 | 1.013 | 7.00 × 10−3 | DNA | RefSeq Genes | TCGA 2 |
| CACNA1B | 582 | 37 | 18,823 | 1.019 | 1.00 × 10−3 | DNA | RefSeq Genes | TCGA 2 |
| CACNA1G | 582 | 37 | 18,823 | 1.027 | 4.56 × 10−16 | DNA | RefSeq Genes | TCGA 2 |
| Gene | Study Dataset | Number of Patients | p-Value | Concordance Index |
|---|---|---|---|---|
| CTTN | Nutt Louis [42] | 50 | 2.12 × 10−5 | 65.39 |
| CTTN+CACNA1F | Nutt Louis [42] | 50 | 4.49 × 10−6 | 67.25 |
| MMP2 | Freije Nelson GPL96 [43] | 85 | 5.95 × 10−3 | 59.74 |
| MMP2+CACNA1B | Freije Nelson GPL96 [43] | 85 | 1.71 × 10−3 | 62.46 |
| MMP2+CACNA1F | Freije Nelson GPL96 [43] | 85 | 5.05 × 10−3 | 60.94 |
| MMP2+CACNA1S | Freije Nelson GPL96 [43] | 85 | 1.87 × 10−3 | 62.04 |
| MMP2+KCNH2 | Freije Nelson GPL96 [43] | 85 | 4.28 × 10−4 | 64.08 |
| MMP2+KCNJ10 | Freije Nelson GPL96 [43] | 85 | 1.32 × 10−3 | 61.17 |
| MMP2+KCNN4 | Freije Nelson GPL96 [43] | 85 | 2.51 × 10−3 | 61.97 |
| MMP2+SCN8A | Freije Nelson GPL96 [43] | 85 | 1.90 × 10−3 | 61.26 |
| MMP9 | Freije Nelson GPL96 [43] | 85 | 2.18 × 10−3 | 59.84 |
| MMP9+CACNA1B | Freije Nelson GPL96 [43] | 85 | 1.36 × 10−3 | 63.62 |
| MMP9+CACNA1G | Freije Nelson GPL96 [43] | 85 | 4.06 × 10−5 | 65.92 |
| MMP9+KCN5A | Freije Nelson GPL96 [43] | 85 | 1.31 × 10−4 | 63.24 |
| Nck | Freije Nelson GPL96 [43] | 85 | 1.05 × 10−3 | 62.01 |
| Nck+CACNA1C | Freije Nelson GPL96 [43] | 85 | 3.55 × 10−4 | 62.17 |
| Nck+CACNA1G | Freije Nelson GPL96 [43] | 85 | 5.06 × 10−4 | 62.94 |
| Nck+CACNA1I | Freije Nelson GPL96 [43] | 85 | 1.61 × 10−5 | 64.53 |
| Nck+CACNA1S | Freije Nelson GPL96 [43] | 85 | 2.50 × 10−4 | 63.33 |
| Nck+KCNA5 | Freije Nelson GPL96 [43] | 85 | 4.68 × 10−4 | 62.1 |
| Nck+KCNH2 | Freije Nelson GPL96 [43] | 85 | 2.27 × 10−4 | 63.92 |
| Nck+KCNJ10 | Freije Nelson GPL96 [43] | 85 | 1.35 ×10−4 | 62.56 |
| SH3PXD2A | Yamanaka Nishio [38] | 29 | 3.80 × 10−2 | 77.89 |
| SH3PXD2A+KCNA5 | Yamanaka Nishio [38] | 29 | 1.49 × 10−2 | 84.21 |
| SH3PXD2A+KCNJ10 | Yamanaka Nishio [38] | 29 | 1.04 × 10−3 | 84.21 |
| SH3PXD2A+SCN5A | Yamanaka Nishio [38] | 29 | 7.27 × 10−3 | 85.26 |
| Src | Lee Nelson GPL570 [40] | 27 | 3.80 × 10−2 | 62.36 |
| Src+CACNA1D | Lee Nelson GPL570 [40] | 27 | 2.10 × 10−2 | 64.94 |
| Src+CACNA1G | Lee Nelson GPL570 [40] | 27 | 3.04 × 10−2 | 58.91 |
| Src+CACNA1S | Lee Nelson GPL570 [40] | 27 | 3.39 × 10−2 | 61.21 |
| Src+KCNB1 | Lee Nelson GPL570 [40] | 27 | 3.39 × 10−2 | 61.78 |
| Src+SCN8A | Lee Nelson GPL570 [40] | 27 | 2.10 × 10−2 | 60.92 |
| Src | Nutt Louis [42] | 50 | 1.10 × 10−2 | 56.48 |
| Src+CACNA1F | Nutt Louis [42] | 50 | 4.10 × 10−3 | 62.49 |
| Src+CACNA1H | Nutt Louis [42] | 50 | 6.58 × 10−3 | 65.8 |
| Src | GBM TCGA | 538 | 5.76 × 10−3 | 54.65 |
| Src+CACNA1C | GBM TCGA | 538 | 6.60 × 10−3 | 54.68 |
| Src+CACNA1G | GBM TCGA | 538 | 7.24 × 10−3 | 54.67 |
| Src+CACNA1H | GBM TCGA | 538 | 7.75 × 10−3 | 54.59 |
| Src+KCNA5 | GBM TCGA | 538 | 7.48 × 10−3 | 54.59 |
| Src+KCNN4 | GBM TCGA | 538 | 7.03 × 10−3 | 54.82 |
| Drug | Indication | Ion Channel |
|---|---|---|
| Amiloride hydrochloride dihydrate | Cardiovascular disease | Sodium |
| Ouabain | Neurological disease | Sodium |
| Oxcarbazepine | Neurological disease | Sodium |
| Primidone | Neurological disease | Sodium |
| Procaine hydrochloride | Neurological disease | Sodium |
| Zonisamide | Neurological disease | Sodium |
| Azelnidipine | Neurological disease | Calcium |
| Cinepazide maleate | Inflammation | Calcium |
| Diltiazem hydrochloride | Cardiovascular disease | Calcium |
| Econazole nitrate | Neurological disease | Calcium |
| Flunarizine dihydrochloride | Neurological disease | Calcium |
| Nicardipine hydrochloride | Neurological disease | Calcium |
| Nilvadipine | Cardiovascular disease | Calcium |
| Glimepiride | Type 2 diabetes mellitus | Potassium |
| Glyburide | Endocrinology | Potassium |
| Nateglinide | Immunology | Potassium |
| Quinine hydrochloride dihydrate | Cardiovascular disease | Potassium |
| Repaglinide | Endocrinology | Potassium |
| Tolbutamide | Type 2 diabetes mellitus | Potassium |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinevska, M.; Gazibegovic, N.; Morokoff, A.P.; Kaye, A.H.; Drummond, K.J.; Mantamadiotis, T.; Stylli, S.S. Inhibition of Radiation and Temozolomide-Induced Glioblastoma Invadopodia Activity Using Ion Channel Drugs. Cancers 2020, 12, 2888. https://doi.org/10.3390/cancers12102888
Dinevska M, Gazibegovic N, Morokoff AP, Kaye AH, Drummond KJ, Mantamadiotis T, Stylli SS. Inhibition of Radiation and Temozolomide-Induced Glioblastoma Invadopodia Activity Using Ion Channel Drugs. Cancers. 2020; 12(10):2888. https://doi.org/10.3390/cancers12102888
Chicago/Turabian StyleDinevska, Marija, Natalia Gazibegovic, Andrew P. Morokoff, Andrew H. Kaye, Katharine J. Drummond, Theo Mantamadiotis, and Stanley S. Stylli. 2020. "Inhibition of Radiation and Temozolomide-Induced Glioblastoma Invadopodia Activity Using Ion Channel Drugs" Cancers 12, no. 10: 2888. https://doi.org/10.3390/cancers12102888
APA StyleDinevska, M., Gazibegovic, N., Morokoff, A. P., Kaye, A. H., Drummond, K. J., Mantamadiotis, T., & Stylli, S. S. (2020). Inhibition of Radiation and Temozolomide-Induced Glioblastoma Invadopodia Activity Using Ion Channel Drugs. Cancers, 12(10), 2888. https://doi.org/10.3390/cancers12102888