Aggressive NK Cell Leukemia: Current State of the Art



Abstract
:Simple Summary
Abstract
1. Introduction
2. Historical Overview
3. Clinical Features
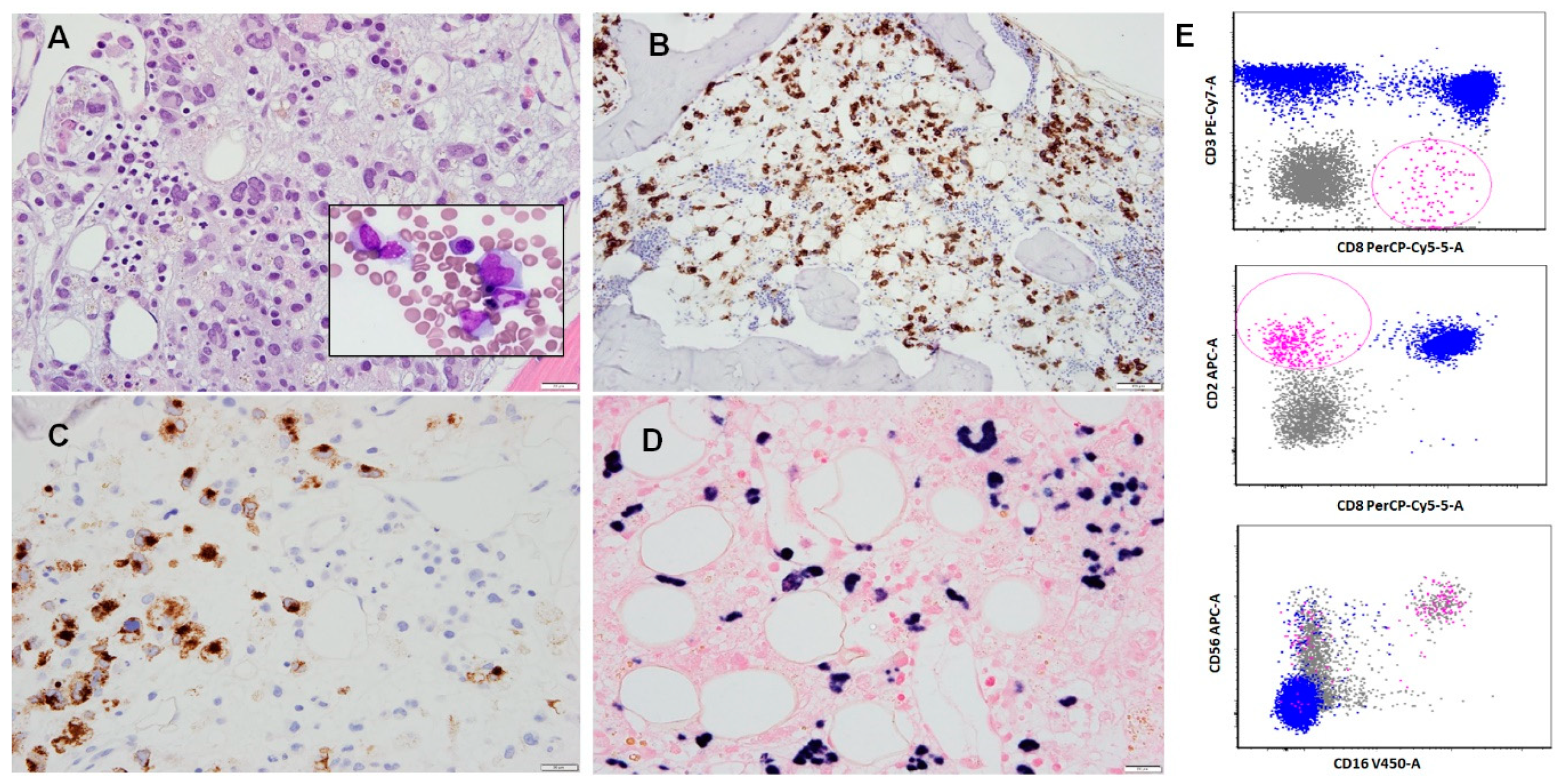
4. Morphologic Features
5. Immunophenotypic Features
6. The Role of Epstein–Barr Virus
7. Cytogenetic Features
8. Molecular Pathogenesis and Genomic Landscape
8.1. JAK/STAT Signaling Pathway
8.2. Epigenetic Dysregulation
8.3. TP53 Alterations and DNA Repair
9. Examples of Clinicogenomic Data Integration
10. Immune Checkpoint Status
11. Treatment of ANKL
11.1. Chemotherapy in ANKL
11.2. Allogeneic Hematopoietic Cell Transplantation in ANKL
11.3. Novel Therapeutic Applications
11.3.1. BCL2 Inhibitors
11.3.2. Heat Shock Protein 90 (HSP90) Inhibitors
11.3.3. Other Approaches
12. Conclusions
Funding
Conflicts of Interest
References
- Chan, J.K.C.; Jaffe, E.S.; Ko, Y.H. Aggressive NK-Cell Leukaemia. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2017; pp. 353–354. [Google Scholar]
- Choi, Y.L.; Makishima, H.; Ohashi, J.; Yamashita, Y.; Ohki, R.; Koinuma, K.; Ota, J.; Isobe, Y.; Ishida, F.; Oshimi, K.; et al. DNA microarray analysis of natural killer cell-type lymphoproliferative disease of granular lymphocytes with purified CD3-CD56+ fractions. Leukemia 2004, 18, 556–565. [Google Scholar] [PubMed] [Green Version]
- Epling-Burnette, P.K.; Bai, F.; Wei, S.; Chaurasia, P.; Painter, J.S.; Olashaw, N.; Hamilton, A.; Sebti, S.; Djeu, J.Y.; Loughran, T.P. ERK couples chronic survival of NK cells to constitutively activated Ras in lymphoproliferative disease of granular lymphocytes (LDGL). Oncogene 2004, 23, 9220–9229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scquizzato, E.; Teramo, A.; Miorin, M.; Facco, M.; Piazza, F.; Noventa, F.; Trentin, L.; Agostini, C.; Zambello, R.; Semenzato, G. Genotypic evaluation of killer immunoglobulin-like receptors in NK-type lymphoproliferative disease of granular lymphocytes. Leukemia 2007, 21, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambello, R.; Falco, M.; Della Chiesa, M.; Trentin, L.; Carollo, D.; Castriconi, R.; Cannas, G.; Carlomagno, S.; Cabrelle, A.; Lamy, T.; et al. Expression and function of KIR and natural cytotoxicity receptors in NK-type lymphoproliferative diseases of granular lymphocytes. Blood 2003, 102, 1797–1805. [Google Scholar] [PubMed]
- Fernandez, L.A.; Pope, B.; Lee, C.; Zayed, E. Aggressive natural killer cell leukemia in an adult with establishment of an NK cell line. Blood 1986, 67, 925–930. [Google Scholar]
- Lmamura, N.; Kusunoki, Y.; Kajihara, H.; Okada, K.; Kuramoto, A. Aggressive Natural Killer Cell Leukemia/Lymphoma with N901-Positive: Surface Phenotype: Evidence for the Existence of a Third Lineage in Lymphoid Cells. Acta Haematol. 1988, 80, 121–128. [Google Scholar] [CrossRef]
- Koizumi, S.; Seki, H.; Tachinami, T.; Taniguchi, M.; Matsuda, A.; Taga, K.; Nakarai, T.; Kato, E.; Taniguchi, N.; Nakamura, H. Malignant clonal expansion of large granular lymphocytes with a Leu-11+, Leu-7- surface phenotype: In vitro responsiveness of malignant cells to recombinant human interleukin 2. Blood 1986, 68, 1065–1073. [Google Scholar]
- Kotani, H.; Ishikura, H.; Hayashi, H.; Tsunematsu, T. Aggressive LGL leukemia associated with recurrent fever and gastrointestinal ulcer. Acta Haematol. Jpn. 1988, 51, 272. [Google Scholar]
- Ohno, Y.; Kamezaki, H.A.; Imanaka, H.; Takahashi, T.; Arita, Y.; Amakawa, H.; Uchiyama, R.; Fukuhara, T.; Kita, S. T3-, Leull+, Leu7- LGL leukemia with germline configuration of TcR gene. Acta Haematol. Jpn. 1987, 50, 338. [Google Scholar]
- Takiguchi, T.; Yoshioka, R.; Tachibana, J.; Maekawa, H.; Hirose, Y.; Konda, S.; Konishi, F. NK cell leukemia and T-y lymphocytosis. In New Diagnostic Technique for Leukemia; Ishiyake Publishers: Tokyo, Japan, 1987; pp. 89–99. [Google Scholar]
- Imamura, N.; Kusunoki, Y.; Kawa-Ha, K.; Yumura, K.; Hara, J.; Oda, K.; Abe, K.; Dohy, H.; Inada, T.; Kajihara, H.; et al. Aggressive natural killer cell leukaemia/lymphoma: Report of four cases and review of the literature Possible Existence of a New Clinical Entity Originating From the Third Lineage of Lymphoid Cells. Br. J. Haematol. 1990, 75, 49–56. [Google Scholar] [CrossRef]
- Soler, J.; Bordes, R.; Ortouno, F.; Montagud, M.; Martorell, J.; Pons, C.; Nomdedeu, J.; Lopez-Lopez, J.J.; Prat, J.; Rutllant, M. Aggressive natural killer cell leukaemia/lymphoma in two patients with lethal midline granuloma. Br. J. Haematol. 1994, 86, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Yamashita, Y.; Tsuzuki, T.; Nakayama, A.; Nakazawa, M.; Hasegawa, Y.; Kojima, H.; Nagasawa, T. Lymphomatous features of aggressive NK cell leukaemia/lymphoma with massive necrosis, haemophagocytosis and EB virus infection. Histopathology 2000, 37, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Brody, J.; Susin, M.; Teichberg, S.; Koduru, P.; Urmacher, C.; Hajdu, S.I.; Marino, J.; Hall, W.W. Aggressive Natural Killer Cell Lymphoma/Leukemia: A recently recognized clinicopathologic entity. Am. J. Surg. Pathol. 1993, 17, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; Jaffe, E.S. Commentary: Aggressive NK cell lymphomas: Insights into the spectrum of NK cell derived malignancies. Histopathology 2000, 37, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.C.; Jaffe, E.; Ralfkiaer, E.; Ko, Y.H. Aggressive NK-Cell Leukemia. In World Health Organization Classification of Tumors, Tumors of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2008; pp. 197–200. [Google Scholar]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hussein, S.; Patel, K.P.; Fang, H.; Thakral, B.; Loghavi, S.; Kanagal-Shamanna, R.; Konoplev, S.; Jabbour, E.J.; Medeiros, L.J.; Khoury, J.D. Genomic and Immunophenotypic Landscape of Aggressive NK-Cell Leukemia. Am. J. Surg. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Suzumiya, J.; Nakamura, S.; Aoki, S.; Notoya, A.; Ozaki, S.; Gondo, H.; Hino, N.; Mori, H.; Sugimori, H.; et al. Aggressive natural killer-cell leukemia revisited: Large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia 2004, 18, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Song, S.-Y.; Kim, W.S.; Ko, Y.H.; Kim, K.; Lee, M.H.; Park, K. Aggressive natural killer cell leukemia: Clinical features and treatment outcome. Haematologica 2002, 87, 1343–1345. [Google Scholar]
- Tang, Y.-T.; Wang, D.; Luo, H.; Xiao, M.; Zhou, H.-S.; Liu, D.; Ling, S.-P.; Wang, N.; Hu, X.-L.; Luo, Y.; et al. Aggressive NK-cell leukemia: Clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J. 2017, 7, 660. [Google Scholar] [CrossRef]
- Ishida, F. Aggressive NK-Cell Leukemia. Front. Pediatr. 2018, 6, 292. [Google Scholar] [CrossRef] [Green Version]
- Ruskova, A.; Thula, R.; Chan, G. Aggressive Natural Killer-Cell Leukemia: Report of five cases and review of the literature. Leuk. Lymphoma 2004, 45, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.; Wang, X.; Bao, L.; Gross, S.A.; Hua, F.; Irons, R.D. Aggressive natural killer cell leukemia: Report of a Chinese series and review of the literature. Int. J. Hematol. 2007, 85, 18–25. [Google Scholar] [CrossRef]
- Zhang, Q.; Jing, W.; Ouyang, J.; Zeng, H.; George, S.K.; Liu, Z. Six cases of aggressive natural killer-cell leukemia in a Chinese population. Int. J. Clin. Exp. Pathol. 2014, 7, 3423–3431. [Google Scholar] [PubMed]
- Li, C.; Tian, Y.; Wang, J.; Zhu, L.; Huang, L.; Wang, N.; Xu, D.; Cao, Y.; Li, J.; Zhou, J. Abnormal immunophenotype provides a key diagnostic marker: A report of 29 cases of de novo aggressive natural killer cell leukemia. Transl. Res. 2014, 163, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Luetke-Eversloh, M.; Killig, M.; Romagnani, C. Signatures of human NK cell development and terminal differentiation. Front. Immunol. 2013, 4, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, A.; Michel, T.; Theresine, M.; Andres, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.G.; Jin, Y.M.; Niu, Q.; Zeng, T.T.; Su, J.; Zhu, H.L. Flow cytometric immunophenotyping is of great value to diagnosis of natural killer cell neoplasms involving bone marrow and peripheral blood. Ann. Hematol. 2013, 92, 89–96. [Google Scholar] [CrossRef]
- Kurt, H.; Jorgensen, J.L.; Amin, H.M.; Patel, K.P.; Wang, S.A.; Lin, P.; Kanagal-Shamanna, R.; Loghavi, S.; Thakral, B.; Khogeer, H.A.; et al. Chronic lymphoproliferative disorder of NK-cells: A single-institution review with emphasis on relative utility of multimodality diagnostic tools. Eur. J. Haematol. 2018, 100, 444–454. [Google Scholar] [CrossRef]
- Borrego, F.; Masilamani, M.; Kabat, J.; Sanni, T.B.; Coligan, J.E. The cell biology of the human natural killer cell CD94/NKG2A inhibitory receptor. Mol. Immunol. 2005, 42, 485–488. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Arlettaz, L.; Villard, J.; de Rham, C.; Degermann, S.; Chapuis, B.; Huard, B.; Roosnek, E. Activating CD94:NKG2C and inhibitory CD94:NKG2A receptors are expressed by distinct subsets of committed CD8+ TCR alphabeta lymphocytes. Eur. J. Immunol. 2004, 34, 3456–3464. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, C.; Biassoni, R.; Pende, D.; Sivori, S.; Accame, L.; Pareti, L.; Semenzato, G.; Moretta, L.; Moretta, A.; Bottino, C. The activating form of CD94 receptor complex: CD94 covalently associates with the Kp39 protein that represents the product of the NKG2-C gene. Eur. J. Immunol. 1998, 28, 327–338. [Google Scholar] [CrossRef]
- Lima, M.; Spinola, A.; Fonseca, S.; Santos, A.H.; Rodrigues, J.; Oliveira, L.; Queiros, M.L.; Santos, M.; Goncalves, M.; Lau, C.; et al. Aggressive mature natural killer cell neoplasms: Report on a series of 12 European patients with emphasis on flow cytometry based immunophenotype and DNA content of neoplastic natural killer cells. Leuk. Lymphoma 2015, 56, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Imamura, T.; Mizunoe, S.; Ohtsuka, E.; Kikuchi, H.; Nasu, M. Natural killer cell-type body cavity lymphoma following chronic active Epstein-Barr virus infection. Am. J. Hematol. 2003, 73, 126–130. [Google Scholar] [CrossRef]
- Lima, M.; Goncalves, C.; Teixeira, M.A.; Franca, M.; Canelhas, A.; Pina, R.; Lopes, V.; Queiros, M.L.; Fonseca, S.; Santos, A.H.; et al. Aggressive natural-killer cell lymphoma presenting with skin lesions, breast nodule, suprarenal masses and life-threatening pericardial and pleural effusions. Leuk. Lymphoma 2001, 42, 1385–1391. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Medeiros, L.J.; Brynes, R.K. Body cavity-based presentation of natural killer cell lymphoma. Leuk. Lymphoma 2005, 46, 293–296. [Google Scholar] [CrossRef]
- Lepeak, L.M.; Yang, D.T.; Chang, J.E. Extranodal NK/T-cell lymphoma presenting with primary cardiac involvement. Hematol. Rep. 2011, 3, e9. [Google Scholar] [CrossRef] [Green Version]
- Kawa-Ha, K.; Ishihara, S.; Ninomiya, T.; Yumura-Yagi, K.; Hara, J.; Murayama, F.; Tawa, A.; Hirai, K. CD3-negative lymphoproliferative disease of granular lymphocytes containing Epstein-Barr viral DNA. J. Clin. Investig. 1989, 84, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Kanegane, H.; Yachie, A.; Miyawaki, T.; Tosato, G. EBV-NK cells interactions and lymphoproliferative disorders. Leuk Lymphoma 1998, 29, 491–498. [Google Scholar] [CrossRef]
- Ng, S.B.; Khoury, J.D. Epstein-Barr virus in lymphoproliferative processes: An update for the diagnostic pathologist. Adv. Anat. Pathol. 2009, 16, 40–55. [Google Scholar] [CrossRef]
- Gao, J.; Behdad, A.; Ji, P.; Wolniak, K.L.; Frankfurt, O.; Chen, Y.H. EBV-negative aggressive NK-cell leukemia/lymphoma: A clinical and pathological study from a single institution. Mod. Pathol. 2017, 30, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, A.; Ganapathi, K.A.; Pham, T.H.; Xi, L.; Torres-Cabala, C.A.; Nanaji, N.M.; Zha, H.D.; Fan, Z.; Irwin, S.; Pittaluga, S.; et al. EBV-negative Aggressive NK-cell Leukemia/Lymphoma: Clinical, Pathologic, and Genetic Features. Am. J. Surg. Pathol. 2017, 41, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitani, A.K.; Khonglah, Y.; Kumar, R.; Gogoi, B.B.; Jajodia, E. Natural Killer Cell Lymphoma: A Case with Classification Dilemma. J. Clin. Diagn. Res. 2016, 10, ED07. [Google Scholar] [CrossRef] [PubMed]
- Matano, S.; Nakamura, S.; Nakamura, S.; Annen, Y.; Hattori, N.; Kobayashi, K.; Kyoda, K.; Sugimoto, T. Monomorphic agranular natural killer cell lymphoma/leukemia with no Epstein-Barr virus association. Acta Haematol. 1999, 101, 206–208. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, Q.; Yin, W.; Xu, L.; Lie, L. Adult aggressive natural killer cell leukemia. Am. J. Med. Sci. 2013, 346, 56–63. [Google Scholar] [CrossRef]
- Park, J.A.; Jun, K.R.; Nam, S.H.; Ghim, T.T. Favorable outcome in a child with EBV-negative aggressive NK cell leukemia. Int. J. Hematol. 2013, 97, 673–676. [Google Scholar] [CrossRef]
- Ko, Y.H.; Park, S.; Kim, K.; Kim, S.J.; Kim, W.S. Aggressive natural killer cell leukemia: Is Epstein-Barr virus negativity an indicator of a favorable prognosis? Acta Haematol. 2008, 120, 199–206. [Google Scholar] [CrossRef]
- Wong, K.F.; Chan, J.K.; Kwong, Y.L. Identification of del (6) (q21q25) as a recurring chromosomal abnormality in putative NK cell lymphoma/leukaemia. Br. J. Haematol. 1997, 98, 922–926. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.F.; Hsu, C.Y.; Ho, D.M. Aggressive natural killer (NK)-cell leukaemia and extranodal NK/T-cell lymphoma are two distinct diseases that differ in their clinical presentation and cytogenetic findings. Histopathology 2018, 72, 955–964. [Google Scholar] [CrossRef]
- Nakashima, Y.; Tagawa, H.; Suzuki, R.; Karnan, S.; Karube, K.; Ohshima, K.; Muta, K.; Nawata, H.; Morishima, Y.; Nakamura, S.; et al. Genome-wide array-based comparative genomic hybridization of natural killer cell lymphoma/leukemia: Different genomic alteration patterns of aggressive NK-cell leukemia and extranodal Nk/T-cell lymphoma, nasal type. Genes Chromosom. Cancer 2005, 44, 247–255. [Google Scholar] [CrossRef]
- Iqbal, J.; Kucuk, C.; Deleeuw, R.J.; Srivastava, G.; Tam, W.; Geng, H.; Klinkebiel, D.; Christman, J.K.; Patel, K.; Cao, K.; et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia 2009, 23, 1139–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucuk, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Park, H.Y.; Kang, S.Y.; Kim, S.J.; Hwang, J.; Lee, S.; Kwak, S.H.; Park, K.S.; Yoo, H.Y.; Kim, W.S.; et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget 2015, 6, 17764–17776. [Google Scholar] [CrossRef] [Green Version]
- Koo, G.C.; Tan, S.Y.; Tang, T.; Poon, S.L.; Allen, G.E.; Tan, L.; Chong, S.C.; Ong, W.S.; Tay, K.; Tao, M.; et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012, 2, 591–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchekioua, A.; Scourzic, L.; de Wever, O.; Zhang, Y.; Cervera, P.; Aline-Fardin, A.; Mercher, T.; Gaulard, P.; Nyga, R.; Jeziorowska, D.; et al. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia 2014, 28, 338–348. [Google Scholar] [CrossRef]
- Huang, Y.; de Reynies, A.; de Leval, L.; Ghazi, B.; Martin-Garcia, N.; Travert, M.; Bosq, J.; Briere, J.; Petit, B.; Thomas, E.; et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood 2010, 115, 1226–1237. [Google Scholar] [CrossRef]
- Karube, K.; Nakagawa, M.; Tsuzuki, S.; Takeuchi, I.; Honma, K.; Nakashima, Y.; Shimizu, N.; Ko, Y.H.; Morishima, Y.; Ohshima, K.; et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood 2011, 118, 3195–3204. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Gu, Z.H.; Yan, Z.X.; Zhao, X.; Xie, Y.Y.; Zhang, Z.G.; Pan, C.M.; Hu, Y.; Cai, C.P.; Dong, Y.; et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat. Genet. 2015, 47, 1061–1066. [Google Scholar] [CrossRef]
- Dufva, O.; Kankainen, M.; Kelkka, T.; Sekiguchi, N.; Awad, S.A.; Eldfors, S.; Yadav, B.; Kuusanmaki, H.; Malani, D.; Andersson, E.I.; et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018, 9, 1567. [Google Scholar] [CrossRef]
- Huang, L.; Liu, D.; Wang, N.; Ling, S.; Tang, Y.; Wu, J.; Hao, L.; Luo, H.; Hu, X.; Sheng, L.; et al. Integrated genomic analysis identifies deregulated JAK/STAT-MYC-biosynthesis axis in aggressive NK-cell leukemia. Cell Res. 2018, 28, 172–186. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.J.; Dai, W.; O’Mara, M.L.; Abankwa, D.; Chhabra, Y.; Pelekanos, R.A.; Gardon, O.; Tunny, K.A.; Blucher, K.M.; Morton, C.J.; et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 2014, 344, 1249783. [Google Scholar] [CrossRef] [PubMed]
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR interaction code: Predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell 2012, 47, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, S.R.; Nelson, E.A.; Yeh, J.E.; Pinello, L.; Yuan, G.C.; Frank, D.A. STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol. Cell Biol. 2013, 33, 2879–2890. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.K.; Oh, J.; Li, P.; West, E.E.; Wong, E.A.; Andraski, A.B.; Spolski, R.; Yu, Z.X.; He, J.; Kelsall, B.L.; et al. The cytokines IL-21 and GM-CSF have opposing regulatory roles in the apoptosis of conventional dendritic cells. Immunity 2013, 38, 514–527. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wei, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Dobashi, A.; Tsuyama, N.; Asaka, R.; Togashi, Y.; Ueda, K.; Sakata, S.; Baba, S.; Sakamoto, K.; Hatake, K.; Takeuchi, K. Frequent BCOR aberrations in extranodal NK/T-Cell lymphoma, nasal type. Genes Chromosom. Cancer 2016, 55, 460–471. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Sugimoto, K.; Kitoh, T.; Sasaki, M.; Mukai, K.; Ando, J.; Egashira, M.; Schuster, S.M.; Oshimi, K. Selective apoptosis of natural killer-cell tumours by l-asparaginase. Br. J. Haematol. 2005, 130, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Ishida, F.; Ko, Y.H.; Kim, W.S.; Suzumiya, J.; Isobe, Y.; Oshimi, K.; Nakamura, S.; Suzuki, R. Aggressive natural killer cell leukemia: Therapeutic potential of L-asparaginase and allogeneic hematopoietic stem cell transplantation. Cancer Sci. 2012, 103, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.S.; Cho, S.H.; Kim, S.J.; Ko, Y.H.; Kang, E.S.; Kim, W.S. L-asparaginase-based regimens followed by allogeneic hematopoietic stem cell transplantation improve outcomes in aggressive natural killer cell leukemia. J. Hematol. Oncol. 2016, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.M.; Zhao, S.; Liu, W.P.; Zhang, W.Y.; Li, G.D.; Kucuk, C.; Hu, X.Z.; Chan, W.C.; Tang, Y.; Ding, W.S.; et al. Clinicopathologic Characterization of Aggressive Natural Killer Cell Leukemia Involving Different Tissue Sites. Am. J. Surg. Pathol. 2016, 40, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Makishima, H.; Nakazawa, H.; Kobayashi, H.; Shimodaira, S.; Nakazawa, Y.; Kitano, K.; Matsuda, K.; Hidaka, E.; Ishida, F. Promising approach for aggressive NK cell leukaemia with allogeneic haematopoietic cell transplantation. Eur. J. Haematol. 2008, 81, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Hamadani, M.; Kanate, A.S.; DiGilio, A.; Ahn, K.W.; Smith, S.M.; Lee, J.W.; Ayala, E.; Chao, N.; Hari, P.; Bolanos-Meade, J.; et al. Allogeneic Hematopoietic Cell Transplantation for Aggressive NK Cell Leukemia. A Center for International Blood and Marrow Transplant Research Analysis. Biol. Blood Marrow Transplant. 2017, 23, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gonen, M.; Gozman, A.; et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Investig. 2010, 120, 3578–3593. [Google Scholar] [CrossRef]
- Nagel, S.; Venturini, L.; Marquez, V.E.; Meyer, C.; Kaufmann, M.; Scherr, M.; MacLeod, R.A.; Drexler, H.G. Polycomb repressor complex 2 regulates HOXA9 and HOXA10, activating ID2 in NK/T-cell lines. Mol. Cancer 2010, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Ng, S.B.; Tay, J.L.; Lin, B.; Koh, T.L.; Tan, J.; Selvarajan, V.; Liu, S.C.; Bi, C.; Wang, S.; et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 2013, 121, 4512–4520. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Yang, W.I.; Min, Y.H.; Ko, Y.H.; Yoon, S.O. The role of the polycomb repressive complex pathway in T and NK cell lymphoma: Biological and prognostic implications. Tumour. Biol. 2016, 37, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Bullinger, L.; Klapproth, K.; Fiedler, K.; Kestler, H.A.; Barth, T.F.; Moller, P.; Stilgenbauer, S.; Pollack, J.R.; Wirth, T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 2008, 112, 4202–4212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. Polycomb group proteins and MYC: The cancer connection. Cell Mol. Life Sci. 2014, 71, 257–269. [Google Scholar] [CrossRef]
- Neri, F.; Zippo, A.; Krepelova, A.; Cherubini, A.; Rocchigiani, M.; Oliviero, S. Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol. Cell Biol. 2012, 32, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Yang, C.Y.; Wang, L.; Gao, H.X.; Rakhshandehroo, T.; Afghani, S.; Pincus, L.; Balassanian, R.; Rubenstein, J.; Gill, R.; et al. Cutaneous T cell lymphoma PDX drug screening platform identifies cooperation between inhibitions of PI3Kalpha/delta and HDAC. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]


{kind=link}
| Diagnosis | Etiology | Immunophenotype | Functionality | Genomic Landscape |
|---|---|---|---|---|
| Transient increase in NK cells | Autoimmune disorders, viral infections | |||
| CLPD* | Unknown stimulus, possibly viral | CD8+ (uniform positivity), CD16+, CD56+ (dim); Loss of CD2, CD7 and CD57 | Cytotoxic function | Activating mutations in the STAT3 SH2 domain |
| ENKTL* | Strong association with EBV infection | CD2+, CD56+; Loss of sCD3, CD4, CD8, CD16 and CD57 | Cytokine secretion function | DDX3X, JAK/STAT signaling pathway (STAT3, STAT5B, JAK3, and PTPRK), KIT, CTNNB1, TP53, PRDM1, ATG5, AIM1,FOX03, and HACE1, RAS, MYC, KMT2D/MLL2, ARID1A, EP300, ASXL3, CDKN2A, CDKN2B, CDKN1A, FAS |
| ANKL* | Strong association with EBV infection | CD2+, CD16+, CD56+; Loss of sCD3, CD4, CD7, CD8 and CD57 | Cytokine secretion function | JAK/STAT signaling pathway, TP53, TET2, CREBBP, ASXL1, ASXL2, BRINP3, PRPF40B |
| Consistently Positive Markers | Frequently Positive Markers | Frequently Negative Markers |
|---|---|---|
| CD2 | CD7 | CD3 |
| Cytoplasmic CD3 epsilon | CD8 | CD4 |
| CD16 | CD5 | |
| CD56 | CD57 | |
| CD94 | KIR (CD158a–e) | |
| Granzyme B | T-cell receptors | |
| TIA | ||
| Perforin |
| Mutations | Huang et al. [63] (29 Patients) | Dufva et al. [62] (14 Patients) | El Hussein et al. [19] (6 Patients) |
|---|---|---|---|
| JAK/STAT | STAT3, STAT5B, STAT5A, JAK2, JAK3, STAT6, SOCS1, SOCS3 and PTPN11 (48%) | STAT3 (21%) | JAK1, JAK3, STAT3 (66.6%) |
| RAS/MAPK | -- | (29%) | (16.7%) |
| Epigenetic modifiers | TET2 (28%), CREBBP (21%), KMT2D (21%), BCOR (3%) | SETD2, KMT2D and BCOR (50%), TET2 (7%) | TET2 (16.7%), CREBBP (16.7%), GFI1 (16.7%) |
| RNA helicase (DDX3X) | (7%) | (21%) | -- |
| Cell cycle regulation and DNA damage repair | TP53 (34%) | TP53 (7%) | TP53 (50%), ASXL1 (33.3%), ASXL2 (33.3%), BRINP3 (16.7%) |
| mRNA splicing | -- | -- | PRPF40B (16.7%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hussein, S.; Medeiros, L.J.; Khoury, J.D. Aggressive NK Cell Leukemia: Current State of the Art. Cancers 2020, 12, 2900. https://doi.org/10.3390/cancers12102900
El Hussein S, Medeiros LJ, Khoury JD. Aggressive NK Cell Leukemia: Current State of the Art. Cancers. 2020; 12(10):2900. https://doi.org/10.3390/cancers12102900
Chicago/Turabian StyleEl Hussein, Siba, L. Jeffrey Medeiros, and Joseph D. Khoury. 2020. "Aggressive NK Cell Leukemia: Current State of the Art" Cancers 12, no. 10: 2900. https://doi.org/10.3390/cancers12102900
APA StyleEl Hussein, S., Medeiros, L. J., & Khoury, J. D. (2020). Aggressive NK Cell Leukemia: Current State of the Art. Cancers, 12(10), 2900. https://doi.org/10.3390/cancers12102900