Sphingolipid Pathway as a Source of Vulnerability in IDH1mut Glioma

 ,
,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
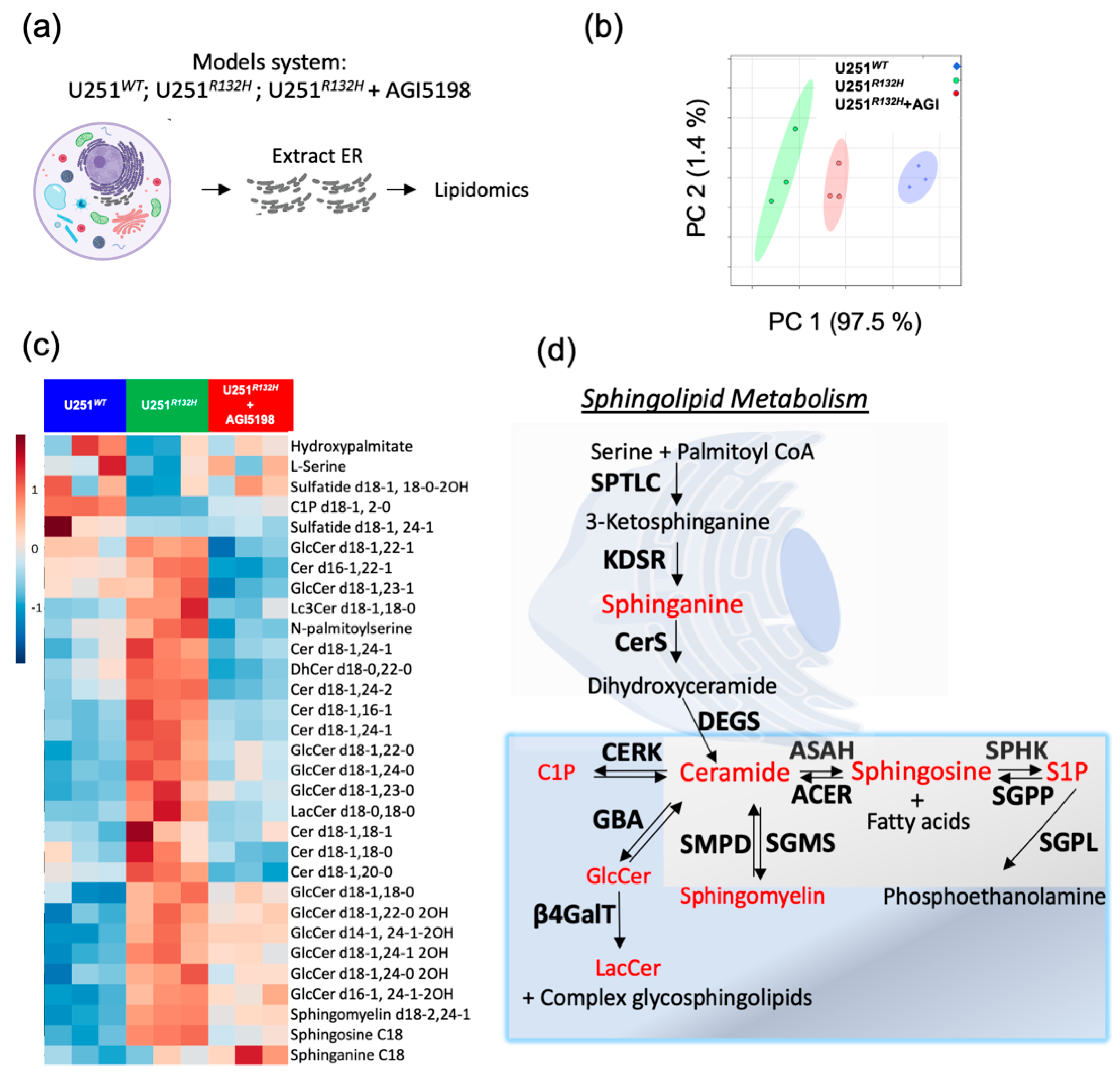
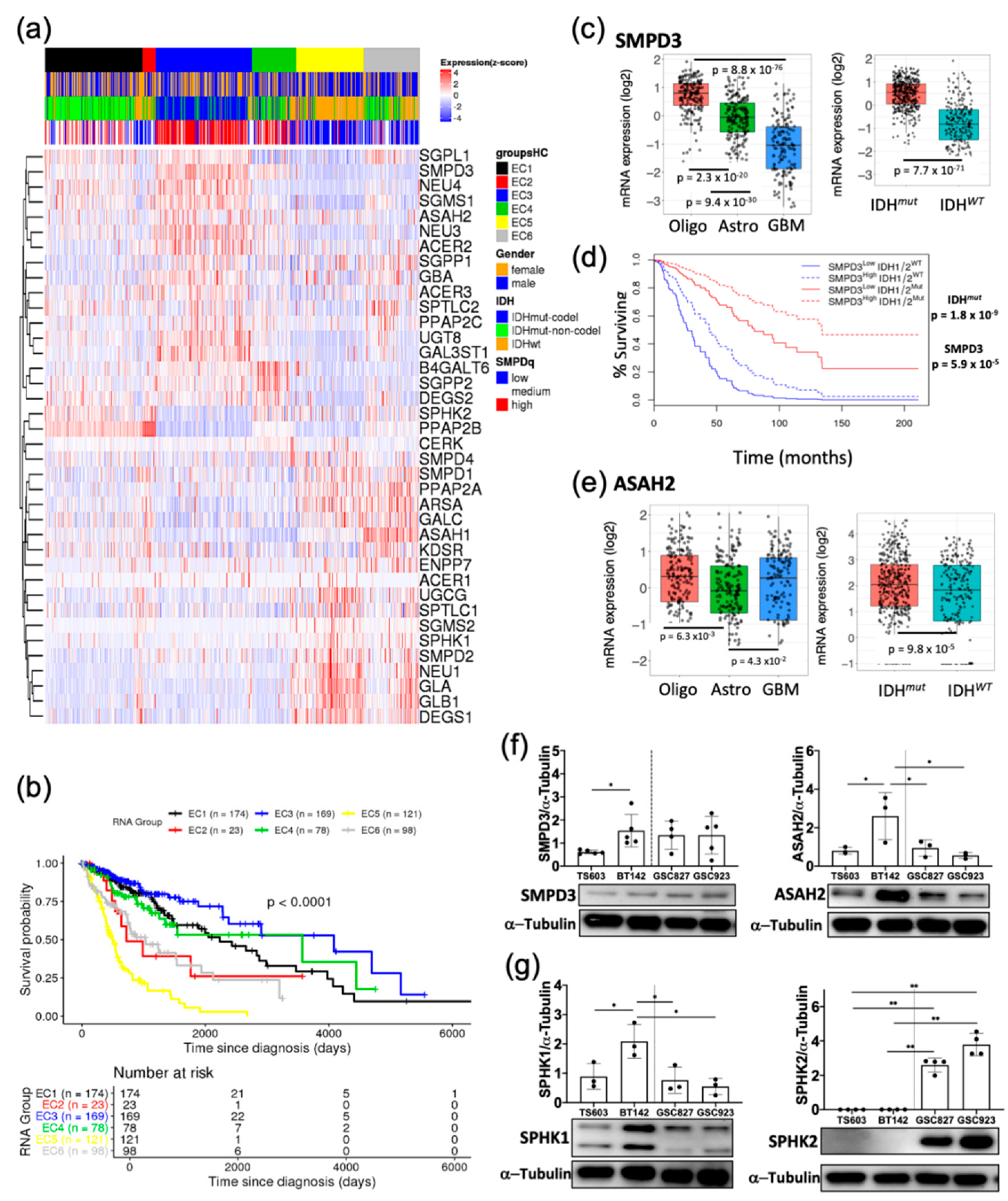
2.1. Sphingolipid Pathway Is Commonly Dysregulated in Astrocytomas and Oligodendroglioma Expressing IDH1-R132H Mutation
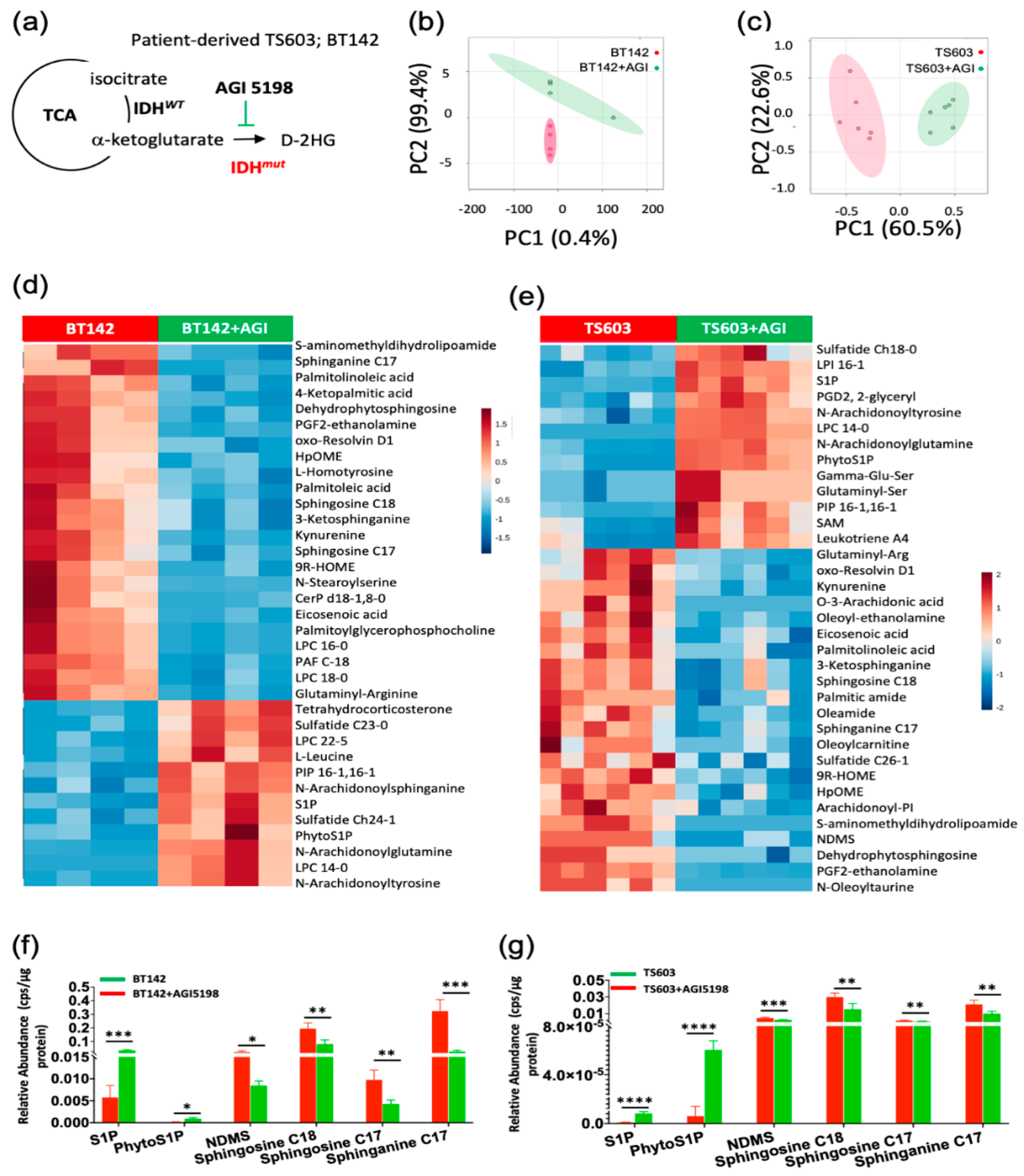
2.2. Probing the Sphingolipid Pathway
2.3. Key Sphingolipid Enzymes are Upregulated IDHmut Patient Samples and Cell Lines
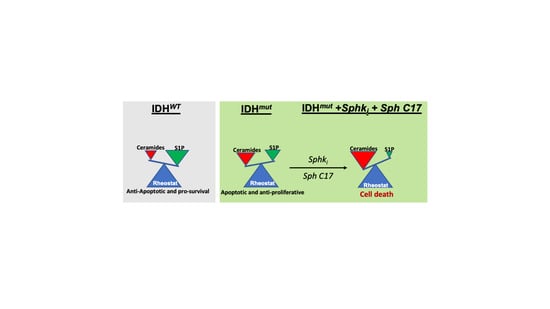
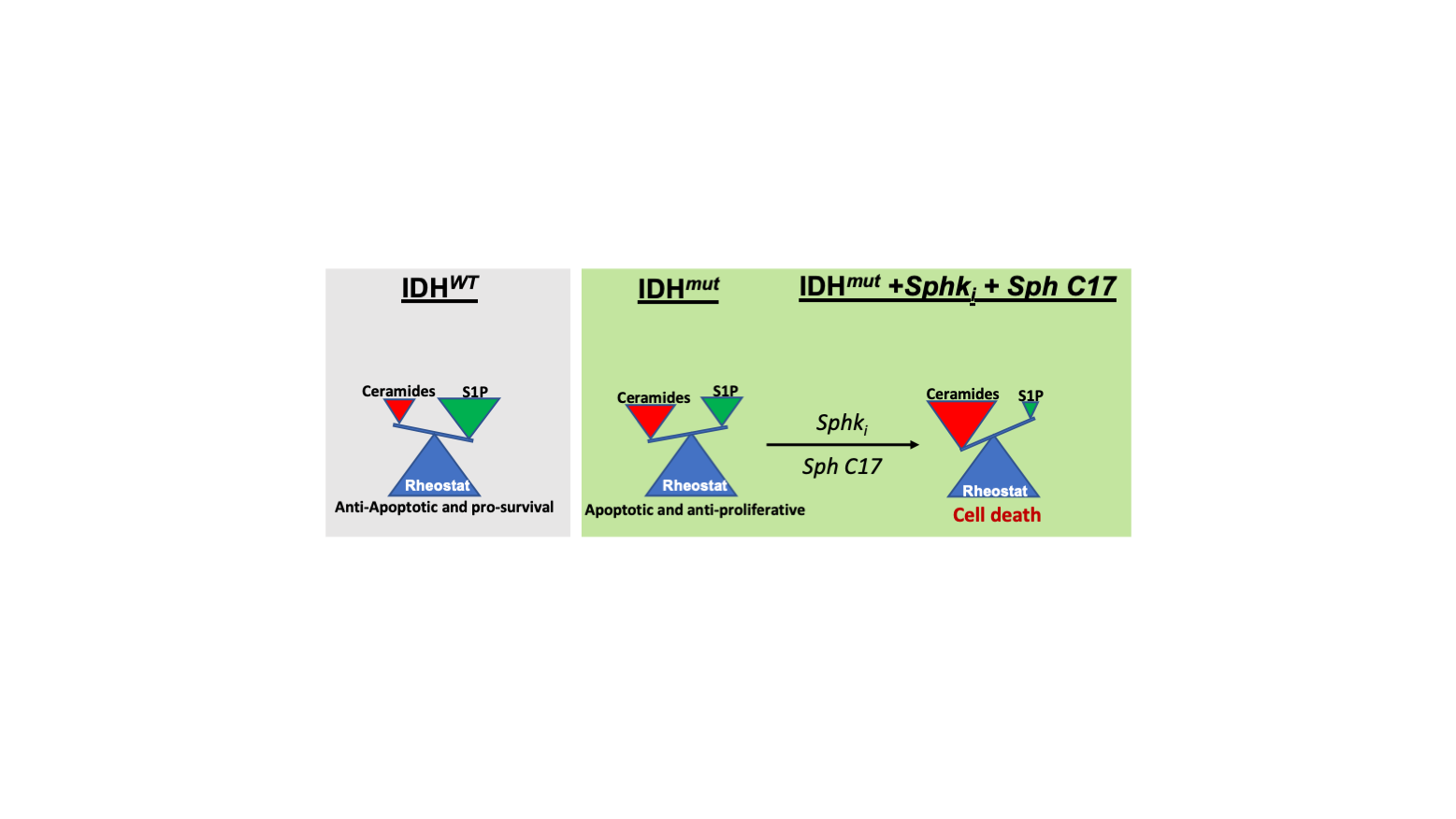
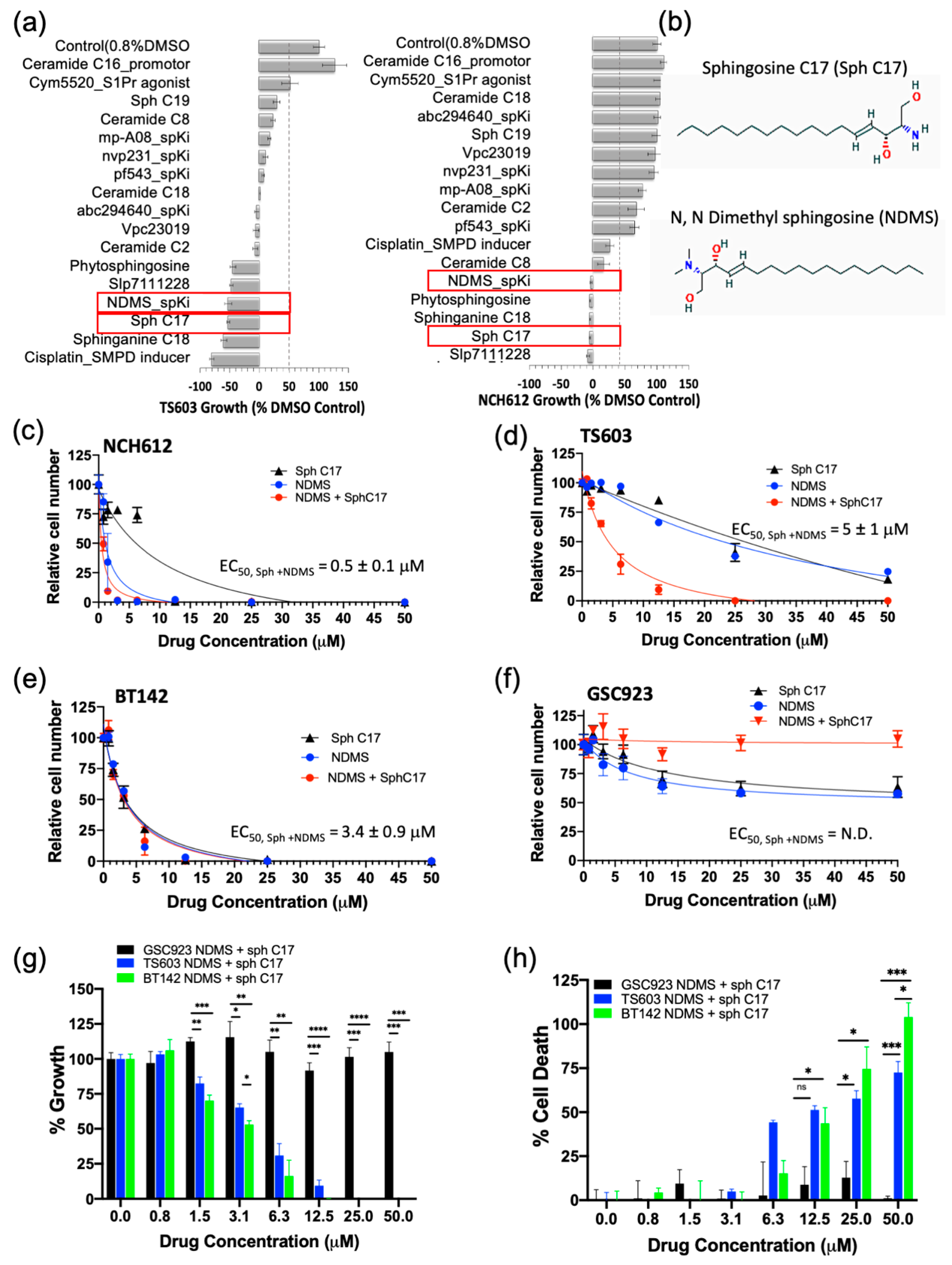
2.4. Targeting the Sphingolipid Pathway in IDH1mut Glioma Cells
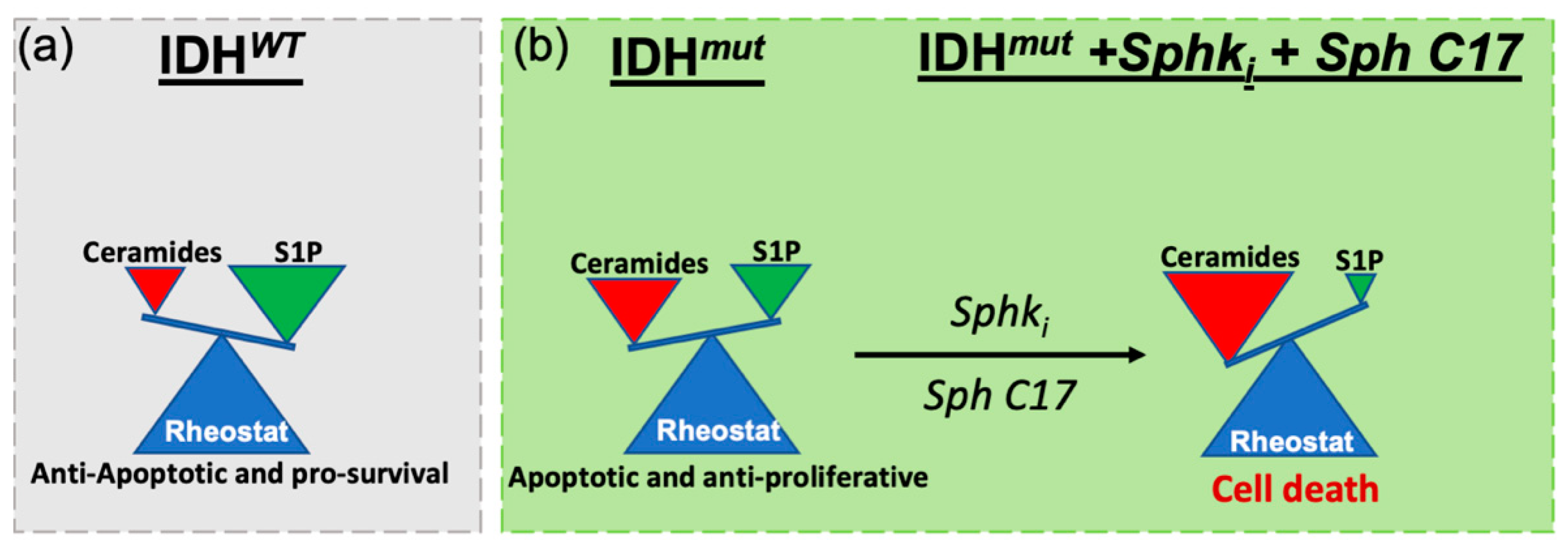
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Sample Collection
4.2. Metabolite Extraction
4.3. Biostatic Drug Screening
4.4. Cell Proliferation Assay (CCK-8)
4.5. LDH Cytotoxicity/Cell Death Assay (CCK-12)
4.6. LC/MS Experimental Set-Up
4.6.1. Organelle Specific Lipidomic Methods for ER
4.6.2. Sphingolipid and Polar Lipid Optimized Methods
4.6.3. LC/MS Data Processing and Statistical Analysis
4.7. TCGA Analysis of Sphingolipid Pathway
4.8. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, Y. Clinical impact of revisions to the WHO classification of diffuse gliomas and associated future problems. Int. J. Clin. Oncol. 2020, 25, 1004–1009. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e125. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Mora, R.; Dokic, I.; Kees, T.; Hüber, C.M.; Keitel, D.; Geibig, R.; Brügge, B.; Zentgraf, H.; Brady, N.R.; Régnier-Vigouroux, A. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia 2010, 58, 1364–1383. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Pistello, M. Sphingolipid/Ceramide Pathways and Autophagy in the Onset and Progression of Melanoma: Novel Therapeutic Targets and Opportunities. Int. J. Mol. Sci. 2019, 20, 3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullmann, T.J.; Siegel, M.I.; Egan, R.W.; Billah, M.M. Sphingosine inhibits phosphatidate phosphohydrolase in human neutrophils by a protein kinase C-independent mechanism. J. Biol. Chem. 1991, 266, 2013–2016. [Google Scholar] [PubMed]
- Meana, C.; García-Rostán, G.; Peña, L.; Lordén, G.; Cubero, Á.; Orduña, A.; Győrffy, B.; Balsinde, J.; Balboa, M.A. The phosphatidic acid phosphatase lipin-1 facilitates inflammation-driven colon carcinogenesis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, S.Y.; Kim, E.H.; Choi, K.S.; Kwon, T.K.; Lee, Y.H.; Chang, J.S.; Kim, M.S.; Jo, Y.H.; Min, D.S. Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol. Cell. Biol. 2003, 23, 3103–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 6, 1603–1624. [Google Scholar] [CrossRef] [Green Version]
- Berdyshev, E.V.; Gorshkova, I.A.; Usatyuk, P.; Zhao, Y.; Saatian, B.; Hubbard, W.; Natarajan, V. De novo biosynthesis of dihydrosphingosine-1-phosphate by sphingosine kinase 1 in mammalian cells. Cell. Signal. 2006, 18, 1779–1792. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lita, A.; Pliss, A.; Kuzmin, A.; Yamasaki, T.; Zhang, L.; Dowdy, T.; Burks, C.; de Val, N.; Celiku, O.; Ruiz-Rodado, V.; et al. IDH1 Mutations Induce Organelle Defects Via Dysregulated Phospholipids. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabalee, J.; Towle, R.; Lawson, J.; Dickman, C.; Garnis, C. Sphingomyelin phosphodiesterase 3 methylation and silencing in oral squamous cell carcinoma results in increased migration and invasion and altered stress response. Oncotarget 2020, 11, 523–534. [Google Scholar] [CrossRef]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.T. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badur, M.G.; Muthusamy, T.; Parker, S.J.; Ma, S.; McBrayer, S.K.; Cordes, T.; Magana, J.H.; Guan, K.L.; Metallo, C.M. Oncogenic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibrosarcoma Sells. Cell Rep. 2018, 25, 1018–1026.e1014. [Google Scholar] [CrossRef] [Green Version]
- Riboni, L.; Abdel Hadi, L.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- Victor, R.R.; Malta, T.M.; Seki, T.; Lita, A.; Dowdy, T.; Celiku, O.; Cavazos-Saldana, A.; Li, A.; Liu, Y.; Han, S.; et al. Metabolic Reprogramming Associated with Aggressiveness Occurs in the G-CIMP-High Molecular Subtypes of IDH1mut Lower Grade Gliomas. Neuro Oncol. 2019. [Google Scholar] [CrossRef]
- Altadill, T.; Dowdy, T.M.; Gill, K.; Reques, A.; Menon, S.S.; Moiola, C.P.; Lopez-Gil, C.; Coll, E.; Matias-Guiu, X.; Cabrera, S.; et al. Metabolomic and Lipidomic Profiling Identifies The Role of the RNA Editing Pathway in Endometrial Carcinogenesis. Sci. Rep. 2017, 7, 8803. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowdy, T.; Zhang, L.; Celiku, O.; Movva, S.; Lita, A.; Ruiz-Rodado, V.; Gilbert, M.R.; Larion, M. Sphingolipid Pathway as a Source of Vulnerability in IDH1mut Glioma. Cancers 2020, 12, 2910. https://doi.org/10.3390/cancers12102910
Dowdy T, Zhang L, Celiku O, Movva S, Lita A, Ruiz-Rodado V, Gilbert MR, Larion M. Sphingolipid Pathway as a Source of Vulnerability in IDH1mut Glioma. Cancers. 2020; 12(10):2910. https://doi.org/10.3390/cancers12102910
Chicago/Turabian StyleDowdy, Tyrone, Lumin Zhang, Orieta Celiku, Sriya Movva, Adrian Lita, Victor Ruiz-Rodado, Mark R. Gilbert, and Mioara Larion. 2020. "Sphingolipid Pathway as a Source of Vulnerability in IDH1mut Glioma" Cancers 12, no. 10: 2910. https://doi.org/10.3390/cancers12102910
APA StyleDowdy, T., Zhang, L., Celiku, O., Movva, S., Lita, A., Ruiz-Rodado, V., Gilbert, M. R., & Larion, M. (2020). Sphingolipid Pathway as a Source of Vulnerability in IDH1mut Glioma. Cancers, 12(10), 2910. https://doi.org/10.3390/cancers12102910