Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer

 , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
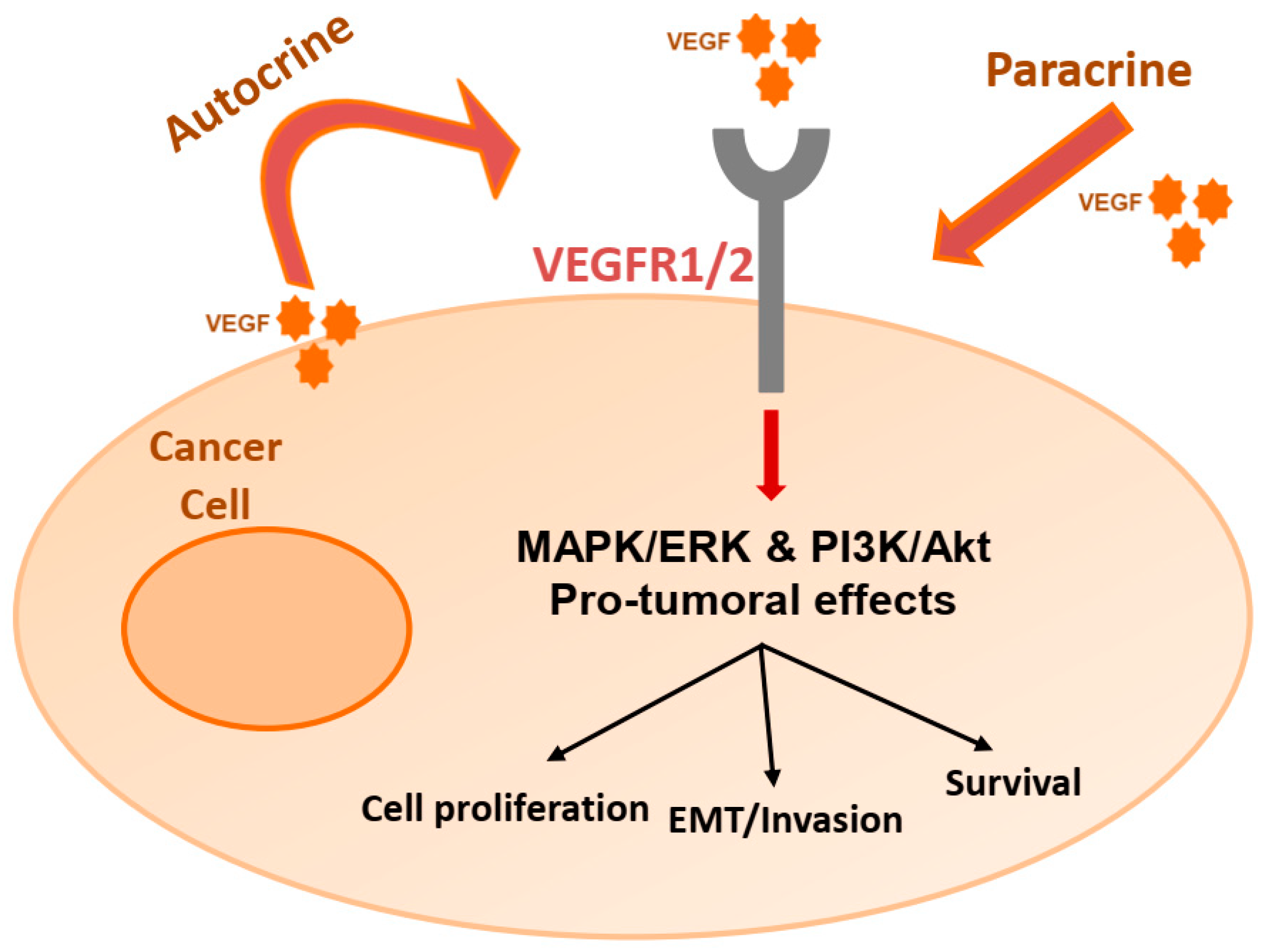
2. The VEGF/VEGFR Pathway
3. Autocrine Effects on Cancer Cells
3.1. Melanoma
3.2. Pancreatic Cancer
3.3. Lung Cancer
3.4. Gastrointestinal Cancer
3.5. Prostate Cancer
3.6. Gliomas
3.7. Breast Cancer
3.8. Hematologic Malignancies
3.9. Other
3.10. VEGF Signaling on Cancer Cells: Stimulation of Survival and Migration
4. Immunomodulatory Effects of the VEGF/VEGFR Pathway
4.1. Immune Cell Infiltration
4.2. Effector T-cells
4.3. Regulatory T-cells (Tregs)
4.4. Dendritic Cells (DCs)
4.5. Myeloid Derived Suppressor Cells (MDSCs)
4.6. Tumor Associated Macrophages (TAMs)
4.7. Combinations of VEGF/VEGFR Inhibition with Cancer Immunotherapy
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Saijo, N.; Tamura, T.; Yamamoto, N.; Nishio, K. New strategies for cancer therapy in the 21st century. Cancer Chemother. Pharmacol. 2001, 48, S102–S106. [Google Scholar] [CrossRef]
- Heron, M. Deaths: Leading Causes for 2017. Nat. Vital Stat. Rep. 2019, 68. Available online: https://www.cdc.gov/nchs/data/nvsr/nvsr68/nvsr68_06-508.pdf?fbclid=IwAR0ShnhUypiDEhFAJvjgxEjArda8ujdLSJj97y3cORXzUHlD_cLPdmzdSdY (accessed on 16 May 2020).
- Ide, A.G.; Baker, N.H.; Warren, S.L. Vascularization of the Brown Pearce rabbit epithelioma transplant as seen in the transparent ear chamber. Am. J. Roentgenol. 1939, 42, 891–899. [Google Scholar]
- Algire, G.H.; Chalkley, H.W.; Legallais, F.Y.; Park, H.D. Vasculae Reactions of Normal and Malignant Tissues in Vivo. I. Vascular Reactions of Mice to Wounds and to Normal and Neoplastic Transplants. J. Natl. Cancer Inst. 1945, 6, 73–85. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 1971, 133, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2019, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nat. Cell Biol. 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Masood, R.; Cai, J.; Zheng, T.; Smith, D.L.; Hinton, D.R.; Gill, P.S. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor–positive human tumors. Blood 2001, 98, 1904–1913. [Google Scholar] [CrossRef]
- Schweighofer, B.; Hofer, E. Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb. Haemost. 2007, 97, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nat. Cell Biol. 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Eklund, L.; Olsen, B.R. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp. Cell Res. 2006, 312, 630–641. [Google Scholar] [CrossRef]
- Héroult, M.; Schaffner, F.; Augustin, H.G. Eph receptor and ephrin ligand-mediated interactions during angiogenesis and tumor progression. Exp. Cell Res. 2006, 312, 642–650. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, F.; Wang, A.; Zheng, S.; Lu, Y. Dll4-Notch signaling in regulation of tumor angiogenesis. J. Cancer Res. Clin. Oncol. 2013, 140, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, E.G.L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- De Vries, C.; Escobedo, J.A.; Ueno, H.; Houck, K.; Ferrara, N.; Williams, L.T. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, K.; Cross, M.J.; Rolny, C.; Hägerkvist, R.; Rahimi, N.; Matsumoto, T.; Claesson-Welsh, L.; Welsh, M.J. The Adaptor Protein Shb Binds to Tyrosine 1175 in Vascular Endothelial Growth Factor (VEGF) Receptor-2 and Regulates VEGF-dependent Cellular Migration. J. Biol. Chem. 2004, 279, 22267–22275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terman, B.I.; Dougher-Vermazen, M.; Carrion, M.E.; Dimitrov, D.; Armellino, D.C.; Gospodarowicz, D.; Böhlen, P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem. Biophys. Res. Commun. 1992, 187, 1579–1586. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Favier, B.; Alam, A.; Barron, P.; Bonnin, J.; Laboudie, P.; Fons, P.; Mandron, M.; Herault, J.P.; Neufeld, G.; Savi, P.; et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006, 108, 1243–1250. [Google Scholar] [CrossRef]
- Pajusola, K.; Aprelikova, O.; Korhonen, J.; Kaipainen, A.; Pertovaara, L.; Alitalo, R.; Alitalo, K. FLT4 receptor tyrosine kinase contains seven immunoglobulin-like loops and is expressed in multiple human tissues and cell lines. Cancer Res. 1992, 52, 5738–5743. [Google Scholar]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nat. Cell Biol. 2005, 438, 946–953. [Google Scholar] [CrossRef]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, M.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nat. Cell Biol. 1996, 380, 435–439. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.-F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nat. Cell Biol. 1995, 376, 62–66. [Google Scholar] [CrossRef]
- Speirs, V.; Atkin, S.L. Production of VEGF and expression of the VEGF receptors Flt-1 and KDR in primary cultures of epithelial and stromal cells derived from breast tumours. Br. J. Cancer 1999, 80, 898–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The Biology of Vascular Endothelial Growth Factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Pentheroudakis, G.; Mavroeidis, L.; Papadopoulou, K.; Koliou, G.-A.; Bamia, C.; Chatzopoulos, K.; Samantas, E.; Mauri, D.; Efstratiou, I.; Pectasides, D.; et al. Angiogenic and Antiangiogenic VEGFA Splice Variants in Colorectal Cancer: Prospective Retrospective Cohort Study in Patients Treated With Irinotecan-Based Chemotherapy and Bevacizumab. Clin. Color. Cancer 2019, 18, e370–e384. [Google Scholar] [CrossRef]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D.O. Molecular Diversity of VEGF-A as a Regulator of Its Biological Activity. Microcirculation 2009, 16, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, C.; Guyot, M.; Pagès, G. VEGF Spliced Variants: Possible Role of Anti-Angiogenesis Therapy. J. Nucleic Acids 2011, 2012, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hellström, M.; Phng, L.-K.; Gerhardt, H. VEGF and Notch Signaling. Cell Adhes. Migr. 2007, 1, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Phng, L.-K.; Gerhardt, H. Angiogenesis: A Team Effort Coordinated by Notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by Vascular Endothelial Growth Factor in Arterial Endothelial Cells: Implications for Modulating Arteriogenesis and Angiogenesis. Mol. Cell. Biol. 2003, 23, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Lobov, I.B.; Renard, R.A.; Papadopoulos, N.; Gale, N.W.; Thurston, G.; Yancopoulos, G.D.; Wiegand, S.J. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.K.; Li, J.-L.; Murga, M.; Harris, A.L.; Tosato, G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 2006, 107, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Sainson, R.C.; Williams, C.K.; Taylor, J.M.; Shi, W.; Li, J.-L.; Harris, A.L. Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc. Res. 2008, 75, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, J.; Ishiwata, T.; Shen, B.; Kornmann, M.; Korc, M. Concomitant over-expression of vascular endothelial growth factor and its receptors in pancreatic cancer. Int. J. Cancer 2000, 85, 27–34. [Google Scholar] [CrossRef]
- Wu, W.; Shu, X.; Hovsepyan, H.; Mosteller, R.D.; Broek, D. VEGF receptor expression and signaling in human bladder tumors. Oncogene 2003, 22, 3361–3370. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Konishi, I.; Mandai, M.; Kuroda, H.; Komatsu, T.; Nanbu, K.; Sakahara, H.; Mori, T. Expression of vascular endothelial growth factor (VEGF) in epithelial ovarian neoplasms: Correlation with clinicopathology and patient survival, and analysis of serum VEGF levels. Br. J. Cancer 1997, 76, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, W.T. Expression of vascular endothelial growth factor and its receptors in multiple myeloma and other hematopoietic malignancies. Semin. Oncol. 2001, 28, 551–559. [Google Scholar] [CrossRef]
- Olson, T.; Mohanraj, D.; Carson, L.F.; Ramakrishnan, S. Vascular permeability factor gene expression in normal and neoplastic human ovaries. Cancer Res. 1994, 54, 276–280. [Google Scholar]
- Paley, P.J.; Staskus, K.A.; Gebhard, K.; Mohanraj, D.; Twiggs, L.B.; Carson, L.F.; Ramakrishnan, S. Vascular endothelial growth factor expression in early stage ovarian carcinoma. Cancer 1997, 80, 98–106. [Google Scholar] [CrossRef]
- Costache, M.; Ioana, M.; Iordache, S.; Ene, D.; Costache, C.A.; Săftoiu, A. VEGF expression in pancreatic cancer and other malignancies: A review of the literature. Rom. J. Intern. Med. 2015, 53, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Dias, S.; Hattori, K.; Zhu, Z.; Heissig, B.; Choy, M.; Lane, W.; Wu, Y.; Chadburn, A.; Hyjek, E.; Gill, M.; et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J. Clin. Investig. 2000, 106, 511–521. [Google Scholar] [CrossRef]
- Gee, M.F.W.; Tsuchida, R.; Eichler-Jonsson, C.; Das, B.; Baruchel, S.; Malkin, D. Vascular endothelial growth factor acts in an autocrine manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene 2005, 24, 8025–8037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Hurwitz, H. Combinations of Bevacizumab with Cancer Immunotherapy. Cancer J. 2018, 24, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Hamerlik, P.; Lathia, J.D.; Rasmussen, R.; Wu, Q.; Bartkova, J.; Lee, M.; Moudry, P.; Bartek, J.; Fischer, W.; Lukas, J.; et al. Autocrine VEGF–VEGFR2–Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med. 2012, 209, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aesoy, R.; Sanchez, B.C.; Norum, J.H.; Lewensohn, R.; Viktorsson, K.; Linderholm, B. An Autocrine VEGF/VEGFR2 and p38 Signaling Loop Confers Resistance to 4-Hydroxytamoxifen in MCF-7 Breast Cancer Cells. Mol. Cancer Res. 2008, 6, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Rikala, M.-S.; Rysz, J.; Lasota, J.; Wang, Z.-F. Vascular Endothelial Growth Factor Receptor 2 as a Marker for Malignant Vascular Tumors and Mesothelioma. Am. J. Surg. Pathol. 2012, 36, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Song, S.; Wu, J.; Meng, L.; Dong, Z.; Shou, C. Vascular Endothelial Growth Factor: Acting as an Autocrine Growth Factor for Human Gastric Adenocarcinoma Cell MGC803. Biochem. Biophys. Res. Commun. 2001, 286, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Spannuth, W.A.; Nick, A.M.; Jennings, N.B.; Armaiz-Pena, G.N.; Mangala, L.S.; Danes, C.G.; Lin, Y.G.; Merritt, W.M.; Thaker, P.H.; Kamat, A.A.; et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int. J. Cancer 2009, 124, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Earl, H.M.; Baban, D.; Shoaibi, M.; Fabra, A.; Kerr, D.; Seymour, L. Melanoma Cell Lines Express VEGF Receptor KDR and Respond to Exogenously Added VEGF. Biochem. Biophys. Res. Commun. 1995, 217, 721–727. [Google Scholar] [CrossRef]
- Ruffini, F.; D’Atri, S.; Lacal, P.M. Neuropilin-1 expression promotes invasiveness of melanoma cells through vascular endothelial growth factor receptor-2-dependent and -independent mechanisms. Int. J. Oncol. 2013, 43, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Wey, J.S.; Fan, F.; Gray, M.J.; Bauer, T.W.; Mccarty, M.F.; Somcio, R.; Liu, W.; Evans, D.B.; Wu, Y.; Hicklin, D.J.; et al. Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer 2005, 104, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Matkar, P.N.; Singh, K.K.; Rudenko, D.; Kim, Y.J.; Kuliszewski, M.A.; Prud’Homme, G.J.; Hedley, D.W.; Leong-Poi, H. Novel regulatory role of neuropilin-1 in endothelial-to-mesenchymal transition and fibrosis in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 69489–69506. [Google Scholar] [CrossRef] [Green Version]
- Barr, M.P.; Gray, S.G.; Gately, K.; Hams, E.; Fallon, P.G.; Davies, A.M.; Richard, D.J.; Pidgeon, G.P.; O’Byrne, K.J. Vascular endothelial growth factor is an autocrine growth factor, signaling through neuropilin-1 in non-small cell lung cancer. Mol. Cancer 2015, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, S.; Ohsaki, Y.; Nakanishi, K.; Toyoshima, E.; Kikuchi, K. Human small cell lung cancer cells express functional VEGF receptors, VEGFR-2 and VEGFR-3. Lung Cancer 2004, 46, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, R.; Ye, X.-C.; Wang, R.; Ling, X.; McManus, M.; Fan, F.; Boulbès, D.; Ellis, L.M. Intracrine VEGF Signaling Mediates the Activity of Prosurvival Pathways in Human Colorectal Cancer Cells. Cancer Res. 2016, 76, 3014–3024. [Google Scholar] [CrossRef] [Green Version]
- Lesslie, D.P.; Summy, J.M.; Parikh, N.U.; Fan, F.; Trevino, J.G.; Sawyer, T.K.; Metcalf, C.A., III; Shakespeare, W.C.; Hicklin, D.J.; Ellis, L.M.; et al. Vascular endothelial growth factor receptor-1 mediates migration of human colorectal carcinoma cells by activation of Src family kinases. Br. J. Cancer 2006, 94, 1710–1717. [Google Scholar] [CrossRef] [Green Version]
- Fan, F.; Wey, J.S.; Mccarty, M.F.; Belcheva, A.; Liu, W.; Bauer, T.W.; Somcio, R.J.; Wu, Y.; Hooper, A.; Hicklin, D.J.; et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 2005, 24, 2647–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, R.C.; Goldsmith, J.D.; Bachelder, R.E.; Brown, C.; Shibuya, M.; Oettgen, P.; Mercurio, A.M. Flt-1-Dependent Survival Characterizes the Epithelial-Mesenchymal Transition of Colonic Organoids. Curr. Biol. 2003, 13, 1721–1727. [Google Scholar] [CrossRef] [Green Version]
- Bates, R.C.; Pursell, B.M.; Mercurio, A.M. Epithelial-Mesenchymal Transition and Colorectal Cancer: Gaining Insights into Tumor Progression Using LIM 1863 Cells. Cells Tissues Organs 2007, 185, 29–39. [Google Scholar] [CrossRef]
- Zhang, H. Expression of vascular endothelial growth factor and its receptors KDR and Flt-1 in gastric cancer cells. World J. Gastroenterol. 2002, 8, 994–998. [Google Scholar] [CrossRef]
- Jackson, M.W.; Roberts, J.S.; Heckford, S.E.; Ricciardelli, C.; Stahl, J.; Choong, C.; Horsfall, D.J.; Tilley, W.D. A potential autocrine role for vascular endothelial growth factor in prostate cancer. Cancer Res. 2002, 62, 854–859. [Google Scholar]
- Ferrer, F.A.; Miller, L.J.; Lindquist, R.; Kowalczyk, P.; Laudone, V.P.; Albertsen, P.C.; Kreutzer, D.L. Expression of vascular endothelial growth factor receptors in human prostate cancer. Urology 1999, 54, 567–572. [Google Scholar] [CrossRef]
- Szabo, E.; Schneider, H.; Seystahl, K.; Rushing, E.J.; Herting, F.; Weidner, K.M.; Weller, M. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro-Oncol. 2016, 18, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knizetova, P.; Hlobilkova, A.; Vancova, I.; Kalita, O.; Ehrmann, J.; Kolar, Z.; Bartek, J. Autocrine regulation of glioblastoma cell-cycle progression, viability and radioresistance through the VEGF-VEGFR2 (KDR) interplay. Cell Cycle 2008, 7, 2553–2561. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wu, X.; Zhu, J. VEGF Promotes Proliferation of Human Glioblastoma Multiforme Stem-Like Cells through VEGF Receptor 2. Sci. World J. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.J.; Miralem, T.; Jiang, S.; Steinberg, R.; Avraham, H. Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 2001, 12, 129–136. [Google Scholar]
- Schoeffner, D.J.; Matheny, S.L.; Akahane, T.; Factor, V.; Berry, A.; Merlino, G.; Thorgeirsson, U.P. VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab. Investig. 2005, 85, 608–623. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Hantel, P.; Kreienberg, R.; Waltenberger, J. Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis 2005, 8, 197–204. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Crago, A.; Chung, J.; Wendt, M.A.; Shaw, L.M.; Robinson, G.; Mercurio, A.M. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001, 61, 5736–5740. [Google Scholar]
- Bachelder, R.; Wendt, M.; Mercurio, A.M. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002, 62, 7203–7206. [Google Scholar]
- Bachelder, R.; Lipscomb, E.; Lin, X.; Wendt, M.; Chadborn, N.H.; Eickholt, B.J.; Mercurio, A.M. Competing autocrine pathways involving alternative neuropilin-1 ligands regulate chemotaxis of carcinoma cells. Cancer Res. 2003, 63, 5230–5233. [Google Scholar] [PubMed]
- Luo, M.; Hou, L.; Li, J.; Shao, S.; Huang, S.; Meng, D.; Liu, L.; Feng, L.; Xia, P.; Qin, T.; et al. VEGF/NRP-1 axis promotes progression of breast cancer via enhancement of epithelial-mesenchymal transition and activation of NF-κB and β-catenin. Cancer Lett. 2016, 373, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Jiang, G.; Zhang, Y.; Xu, J.; Chen, C.; Zhang, L.; Xu, Z.; Du, X. Effects of RNA interference-mediated NRP-1 silencing on the proliferation and apoptosis of breast cancer cells. Mol. Med. Rep. 2012, 12, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2014, 34, 3107–3119. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Li, C.; Zhao, Y.; Wu, L.; Du, X.; Han, Z. VEGF-A/Neuropilin 1 Pathway Confers Cancer Stemness via Activating Wnt/β-Catenin Axis in Breast Cancer Cells. Cell. Physiol. Biochem. 2017, 44, 1251–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.-M.; Zhu, J.-G.; Gu, L.; Hu, S.-Q.; Wu, H. VEGFR2 Expression in Head and Neck Squamous Cell Carcinoma Cancer Cells Mediates Proliferation and Invasion. Asian Pac. J. Cancer Prev. 2016, 17, 2217–2221. [Google Scholar] [CrossRef] [Green Version]
- Kopparapu, P.K.; Boorjian, S.A.; Robinson, B.D.; Downes, M.; Gudas, L.J.; Mongan, N.P.; Persson, J.L. Expression of VEGF and its receptors VEGFR1/VEGFR2 is associated with invasiveness of bladder cancer. Anticancer Res. 2013, 33, 2381–2390. Available online: http://ar.iiarjournals.org/content/33/6/2381.full (accessed on 18 June 2020).
- Podar, K.; Anderson, K.C. The pathophysiologic role of VEGF in hematologic malignancies: Therapeutic implications. Blood 2005, 105, 1383–1395. [Google Scholar] [CrossRef] [Green Version]
- Podar, K.; Tai, Y.-T.; Davies, F.E.; Lentzsch, S.; Sattler, M.; Hideshima, T.; Lin, B.K.; Gupta, D.; Shima, Y.; Chauhan, D.; et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001, 98, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Fonseca, R.; Greipp, P.R.; Rajkumar, S.V. Expression of VEGF and its receptors by myeloma cells. Leukemia 2003, 17, 2025–2031. [Google Scholar] [CrossRef] [Green Version]
- Vincent, L.; Jin, D.K.; Karajannis, M.A.; Shido, K.; Hooper, A.T.; Rashbaum, W.K.; Pytowski, B.; Wu, Y.; Hicklin, D.J.; Zhu, Z.; et al. Fetal Stromal–Dependent Paracrine and Intracrine Vascular Endothelial Growth Factor-A/Vascular Endothelial Growth Factor Receptor-1 Signaling Promotes Proliferation and Motility of Human Primary Myeloma Cells. Cancer Res. 2005, 65, 3185–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attar-Schneider, O.; Drucker, L.; Zismanov, V.; Tartakover-Matalon, S.; Rashid, G.; Lishner, M. Bevacizumab attenuates major signaling cascades and eIF4E translation initiation factor in multiple myeloma cells. Lab. Investig. 2011, 92, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Lacal, P.M.; Failla, C.M.; Pagani, E.; Odorisio, T.; Schietroma, C.; Falcinelli, S.; Zambruno, G.; D’Atri, S. Human Melanoma Cells Secrete and Respond to Placenta Growth Factor and Vascular Endothelial Growth Factor. J. Investig. Dermatol. 2000, 115, 1000–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagani, E.; Ruffini, F.; Cappellini, G.C.A.; Scoppola, A.; Fortes, C.; Marchetti, P.; Graziani, G.; D’Atri, S.; Lacal, P.M. Placenta growth factor and neuropilin-1 collaborate in promoting melanoma aggressiveness. Int. J. Oncol. 2016, 48, 1581–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Robinson, W.A.; Brady, B.M.; Glode, L.M. Migration and invasion of human prostate cancer cells is related to expression of VEGF and its receptors. Anticancer Res. 2003, 23, 3917–3922. [Google Scholar]
- Mentlein, R.; Forstreuter, F.; Mehdorn, H.M.; Held-Feindt, J. Functional significance of vascular endothelial growth factor receptor expression on human glioma cells. J. Neuro-Oncol. 2004, 67, 9–18. [Google Scholar] [CrossRef]
- Mesti, T.; Savarin, P.; Triba, M.N.; Le Moyec, L.; Ocvirk, J.; Banissi, C.; Carpentier, A.F. Metabolic Impact of Anti-Angiogenic Agents on U87 Glioma Cells. PLoS ONE 2014, 9, e99198. [Google Scholar] [CrossRef] [Green Version]
- Rydén, L.; Linderholm, B.; Nielsen, N.H.; Emdin, S.; Jönsson, P.-E.; Landberg, G. Tumor Specific VEGF-A and VEGFR2/KDR Protein are Co-expressed in Breast Cancer. Breast Cancer Res. Treat. 2003, 82, 147–154. [Google Scholar] [CrossRef]
- Wu, Y.; Hooper, A.T.; Zhong, Z.; Witte, L.; Böhlen, P.; Rafii, S.; Hicklin, D.J. The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int. J. Cancer 2006, 119, 1519–1529. [Google Scholar] [CrossRef]
- Yan, J.-D.; Liu, Y.; Zhang, Z.-Y.; Liu, G.-Y.; Xu, J.-H.; Liu, L.-Y.; Hu, Y.-M. Expression and prognostic significance of VEGFR-2 in breast cancer. Pathol.-Res. Pr. 2015, 211, 539–543. [Google Scholar] [CrossRef]
- Stendahl, M.; Emdin, S.; Bengtsson, N.O. Tumor-specific VEGF-A and VEGFR2 in postmenopausal breast cancer patients with long-term follow-up. Implication of a link between VEGF pathway and tamoxifen response. Breast Cancer Res. Treat. 2005, 89, 135–143. [Google Scholar] [CrossRef]
- Nakopoulou, L.; Stefanaki, K.; Panayotopoulou, E.; Giannopoulou, I.; Athanassiadou, P.; Gakiopoulou-Givalou, H.; Louvrou, A. Expression of the vascular endothelial growth factor receptor-2/Flk-1 in breast carcinomas: Correlation with proliferation. Hum. Pathol. 2002, 33, 863–870. [Google Scholar] [CrossRef]
- Lentzsch, S.; Chatterjee, M.; Gries, M.; Bommert, K.; Gollasch, H.; Dörken, B.; Bargou, R.C. PI3-K/AKT/FKHR and MAPK signaling cascades are redundantly stimulated by a variety of cytokines and contribute independently to proliferation and survival of multiple myeloma cells. Leukemia 2004, 18, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Da Via’, M.C.; Leone, P.; Croci, G.; Borrelli, P.; Gaviria, P.T.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Adhesion-Mediated Multiple Myeloma (MM) Disease Progression: Junctional Adhesion Molecule a Enhances Angiogenesis and Multiple Myeloma Dissemination and Predicts Poor Survival. Blood 2019, 134, 855. [Google Scholar] [CrossRef]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Wang, Y.; Duncan, M.K.; Naik, U.P. Junctional Adhesion Molecule-A Regulates Vascular Endothelial Growth Factor Receptor-2 Signaling-Dependent Mouse Corneal Wound Healing. PLoS ONE 2013, 8, e63674. [Google Scholar] [CrossRef]
- Veronese, L.; Tournilhac, O.; Verrelle, P.; Davi, F.; Dighiero, G.; Chautard, E.; Veyrat-Masson, R.; Kwiatkowski, F.; Goumy, C.; Gouas, L.; et al. Strong correlation between VEGF and MCL-1 mRNA expression levels in B-cell chronic lymphocytic leukemia. Leuk. Res. 2009, 33, 1623–1626. [Google Scholar] [CrossRef]
- Guo, D.; Jia, Q.; Song, H.-Y.; Warren, R.S.; Donner, D.B. Vascular Endothelial Cell Growth Factor Promotes Tyrosine Phosphorylation of Mediators of Signal Transduction That Contain SH2 Domains. J. Biol. Chem. 1995, 270, 6729–6733. [Google Scholar] [CrossRef] [Green Version]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 Is Expressed by Endothelial and Tumor Cells as an Isoform-Specific Receptor for Vascular Endothelial Growth Factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D. Epithelial-mesenchymal transition in morphogenesis, cancer progression and angiogenesis. Exp. Cell Res. 2017, 353, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Marcu, K.B.; Kolettas, E. Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation. Cells 2019, 8, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghersi, G. Roles of molecules involved in epithelial/mesenchymal transition during angiogenesis. Front. Biosci. 2008, 13, 2335–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenta, S.; Zeisberg, E.M.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Holderfield, M.T.; Hughes, C.C.W. Crosstalk Between Vascular Endothelial Growth Factor, Notch, and Transforming Growth Factor-β in Vascular Morphogenesis. Circ. Res. 2008, 102, 637–652. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.; Laskar, S.; Pandey, B. Autocrine IL-8 and VEGF mediate epithelial–mesenchymal transition and invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer cells. Cell. Signal. 2013, 25, 1780–1791. [Google Scholar] [CrossRef]
- Gonzalez-Moreno, O.; Lecanda, J.; Green, J.E.; Segura, V.; Catena, R.; Serrano, D.; Calvo, A. VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Exp. Cell Res. 2010, 316, 554–567. [Google Scholar] [CrossRef]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.-K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of Tumor Invasion and Metastasis by Concurrent Inhibition of c-Met and VEGF Signaling in Pancreatic Neuroendocrine Tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, J.; Liu, B.-R. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vang, K.B.; Vang, K.B.; Castermans, K.; Popescu, F.; Zhang, Y.; Egbrink, M.G.A.O.; Mescher, M.F.; Farrar, M.A.; Griffioen, A.W.; Mayo, K.H. Enhancement of T-cell-Mediated Antitumor Response: Angiostatic Adjuvant to Immunotherapy against Cancer. Clin. Cancer Res. 2011, 17, 3134–3145. [Google Scholar] [CrossRef] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taïeb, J. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-cell Proliferation in Colorectal Cancer. Cancer Res. 2012, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Koinis, F.; Vetsika, E.K.; Aggouraki, D.; Skalidaki, E.; Koutoulaki, A.; Gkioulmpasani, M.; Georgoulias, V.; Kotsakis, A.; Vetsika, E.K.; Georgoulias, V. Effect of First-Line Treatment on Myeloid-Derived Suppressor Cells’ Subpopulations in the Peripheral Blood of Patients with Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, M.; Rovati, B.; Ronzoni, M.; Loupakis, F.; Mariucci, S.; Ricci, V.; Gattoni, E.; Salvatore, L.; Tinelli, C.; Villa, E.; et al. Immunological Effects of Bevacizumab-Based Treatment in Metastatic Colorectal Cancer. Oncology 2010, 79, 187–196. [Google Scholar] [CrossRef]
- Martino, E.; Misso, G.; Pastina, P.; Costantini, S.; Vanni, F.; Gandolfo, C.; Botta, C.; Capone, F.; Lombardi, A.; Pirtoli, L.; et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov. 2016, 2, 16025. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef]
- Osada, T.; Chong, G.; Tansik, R.; Hong, T.; Spector, N.; Kumar, R.; Hurwitz, H.I.; Dev, I.; Nixon, A.B.; Lyerly, H.K.; et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 2008, 57, 1115–1124. [Google Scholar] [CrossRef]
- Alfaro, C.; Suarez, N.; Gonzalez, A.D.L.G.; Solano, S.; Erro, L.; Dubrot, J.; Palazon, A.; Hervasstubbs, S.; Gurpide, A.; Lopez-Picazo, J.M.; et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br. J. Cancer 2009, 100, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [Green Version]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Lawrence, D.; McDermott, D.; Zhou, J.; Rodig, S.; Hodi, F.S. VEGF Neutralization Plus CTLA-4 Blockade Alters Soluble and Cellular Factors Associated with Enhancing Lymphocyte Infiltration and Humoral Recognition in Melanoma. Cancer Immunol. Res. 2016, 4, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finke, J.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib Reverses Type-1 Immune Suppression and Decreases T-Regulatory Cells in Renal Cell Carcinoma Patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.; Stein, M.; Shannon, P.; Eddy, S.; Tyler, A.; Stephenson, J.J.; Catlett, L.; Huang, B.; Healey, D.; Gordon, M. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer 2010, 117, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, O.; Pere, H.; Ravel, P.; Haicheur, N.; Badoual, C.; Merillon, N.; Medioni, J.; Peyrard, S.; Roncelin, S.; Verkarre, V.; et al. A Decrease of Regulatory T Cells Correlates With Overall Survival After Sunitinib-based Antiangiogenic Therapy in Metastatic Renal Cancer Patients. J. Immunother. 2010, 33, 991–998. [Google Scholar] [CrossRef]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib Inhibition of Stat3 Induces Renal Cell Carcinoma Tumor Cell Apoptosis and Reduces Immunosuppressive Cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib Mediates Reversal of Myeloid-Derived Suppressor Cell Accumulation in Renal Cell Carcinoma Patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Van Hooren, L.; Georganaki, M.; Huang, H.; Mangsbo, S.M.; Dimberg, A. Sunitinib enhances the antitumor responses of agonistic CD40-antibody by reducing MDSCs and synergistically improving endothelial activation and T-cell recruitment. Oncotarget 2016, 7, 50277–50289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. 2014, 29, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Ozao-Choy, J.; Ma, G.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.-Y.; Chen, S.-H. The Novel Role of Tyrosine Kinase Inhibitor in the Reversal of Immune Suppression and Modulation of Tumor Microenvironment for Immune-Based Cancer Therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Fang, X.; Gao, Z.; Chen, W.; Tao, F.; Cai, P.; Yuan, H.; Shu, Y.; Xu, Q.; Sun, Y.; et al. Axitinib, a selective inhibitor of vascular endothelial growth factor receptor, exerts an anticancer effect in melanoma through promoting antitumor immunity. Anti-Cancer Drugs 2014, 25, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Du Four, S.; Maenhout, S.K.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Combined VEGFR and CTLA-4 blockade increases the antigen-presenting function of intratumoral DCs and reduces the suppressive capacity of intratumoral MDSCs. Am. J. Cancer Res. 2016, 6, 2514–2531. [Google Scholar] [PubMed]
- Zhao, S.; Ren, S.; Jiang, T.; Zhu, B.; Li, X.; Zhao, C.; Jia, Y.; Shi, J.; Zhang, L.; Liu, X.; et al. Low-dose apatinib optimizes tumor microenvironment and potentiates antitumor effect of PD-1/PD-L1 blockade in lung cancer. Cancer Immunol. Res. 2019, 7, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Shao, J.; Xu, Q.; Meng, F.; Chen, F.; Ding, N.; Du, S.; Zhou, S.; Cai, J.; et al. Immune-Mediated Antitumor Effect By VEGFR2 Selective Inhibitor For Gastric Cancer. OncoTargets Ther. 2019, 12, 9757–9765. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Xu, Y.; Youn, J.-I.; Cabrera, R.; Zhang, X.; Gabrilovich, D.; Nelson, D.R.; Liu, C. Kinase inhibitor Sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab. Investig. 2011, 91, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Nagai, H.; Mukozu, T.; Matsui, D.; Kanekawa, T.; Kanayama, M.; Wakui, N.; Momiyama, K.; Shinohara, M.; Iida, K.; Ishii, K.; et al. Sorafenib Prevents Escape from Host Immunity in Liver Cirrhosis Patients with Advanced Hepatocellular Carcinoma. Clin. Dev. Immunol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Chen, M.-L.; Yan, B.-S.; Lu, W.-C.; Yu, S.-L.; Yang, P.-C.; Cheng, A.-L. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int. J. Cancer 2013, 134, 319–331. [Google Scholar] [CrossRef]
- Hipp, M.M.; Hilf, N.; Walter, S.; Werth, D.; Brauer, K.M.; Radsak, M.P.; Weinschenk, T.; Singh-Jasuja, H.; Brossart, P. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 2008, 111, 5610–5620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkx, A.E.M.; Egbrink, M.G.A.O.; Kuijpers, M.J.E.; Van Der Niet, S.T.; Heijnen, V.V.T.; Steege, J.C.A.B.-T.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of Vascular Endothelial Growth Factor on the Lymphocyte-Endothelium Interactions: Identification of Caveolin-1 and Nitric Oxide as Control Points of Endothelial Cell Anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromp, S.C.; Egbrink, M.G.A.O.; Dings, R.P.M.; Van Velzen, S.; Slaaf, D.W.; Hillen, H.F.P.; Tangelder, G.J.; Reneman, R.S.; Griffioen, A.W. Tumor angiogenesis factors reduce leukocyte adhesion in vivo. Int. Immunol. 2000, 12, 671–676. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Egbrink, M.G.A.O.; Castermans, K.; Van Der Schaft, D.W.J.; Thijssen, V.L.J.L.; Dings, R.P.M.; Kwee, L.; Mayo, K.H.; Wagstaff, J.; Ter Steege, J.C.A.B.; et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006, 20, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Rmili, C.W.; Kiialainen, A.; Kienast, Y.; Mueller, H.-J.; Ooi, C.-H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.J.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Gröne, H.-J.; Hämmerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nat. Cell Biol. 2008, 453, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti–PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Hoerning, A.; Datta, D.; Edelbauer, M.; Stack, M.P.; Calzadilla, K.; Pal, S.; Briscoe, D.M. Cutting Edge: Vascular Endothelial Growth Factor-Mediated Signaling in Human CD45RO + CD4 + T Cells Promotes Akt and ERK Activation and Costimulates IFN-γ Production. J. Immunol. 2009, 184, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-L.; Zhao, H.; Ren, X.-B. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef] [PubMed]
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br. J. Cancer 2012, 107, 1869–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziogas, A.C.; Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Terpos, E.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor Type 2. Int. J. Cancer 2011, 130, 857–864. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nat. Cell Biol. 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Boissel, N.; Rousselot, P.; Raffoux, E.; Cayuela, J.-M.; Maarek, O.; Charron, D.; Degos, L.; Dombret, H.; Toubert, A.; Réa, D. Defective blood dendritic cells in chronic myeloid leukemia correlate with high plasmatic VEGF and are not normalized by imatinib mesylate. Leukemia 2004, 18, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Malugani, F.; Bonfanti, A.; Bucovec, R.; Secondino, S.; Brivio, F.; Ferrari-Bravo, A.; Ferrante, R.; Vigoré, L.; Rovelli, F.; et al. Abnormally enhanced blood concentrations of vascular endothelial growth factor (VEGF) in metastatic cancer patients and their relation to circulating dendritic cells, IL-12 and endothelin-1. J. Boil. Regul. Homeost. Agents 2001, 15, 140–144. [Google Scholar]
- Strauss, L.; Volland, D.; Kunkel, M.; Reichert, T.E. Dual role of VEGF family members in the pathogenesis of head and neck cancer (HNSCC): Possible link between angiogenesis and immune tolerance. Med. Sci. Monit. 2005, 11, BR280–R292. [Google Scholar] [PubMed]
- Della Porta, M.; Danova, M.; Rigolin, G.M.; Brugnatelli, S.; Rovati, B.; Tronconi, C.; Fraulini, C.; Rossi, A.R.; Riccardi, A.; Castoldi, G. Dendritic Cells and Vascular Endothelial Growth Factor in Colorectal Cancer: Correlations with Clinicobiological Findings. Oncology 2005, 68, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Mimura, K.; Kono, K.; Takahashi, A.; Kawaguchi, Y.; Fujii, H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol. Immunother. 2006, 56, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar] [PubMed]
- Dikov, M.M.; Ohm, J.E.; Ray, N.; Tchekneva, E.E.; Burlison, J.; Moghanaki, D.; Nadaf, S.; Carbone, D.P. Differential Roles of Vascular Endothelial Growth Factor Receptors 1 and 2 in Dendritic Cell Differentiation. J. Immunol. 2004, 174, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oussa, N.A.E.; Dahmani, A.; Gomis, M.; Richaud, M.; Andreev, E.; Navab-Daneshmand, A.-R.; Taillefer, J.; Carli, C.; Boulet, S.; Sabbagh, L.; et al. VEGF Requires the Receptor NRP-1 To Inhibit Lipopolysaccharide-Dependent Dendritic Cell Maturation. J. Immunol. 2016, 197, 3927–3935. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-L.; Liang, Y.-C.; Chiang, B.-L. Placental growth factor down-regulates type 1 T helper immune response by modulating the function of dendritic cells. J. Leukoc. Biol. 2007, 82, 1473–1480. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Karakhanova, S.; Link, J.; Heinrich, M.; Shevchenko, I.; Yang, Y.; Hassenpflug, M.; Bunge, H.; Von Ahn, K.; Brecht, R.; Mathes, A.; et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: Importance of myeloid-derived suppressor cells. Oncoimmunology 2015, 4, e998519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Chen, X.; Dikov, M.M.; Novitskiy, S.V.; Mosse, C.A.; Yang, L.; Carbone, D.P. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood 2007, 110, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [Green Version]
- Evoron, T.; Emarcheteau, E.; Epernot, S.; Ecolussi, O.; Tartour, E.; Etaieb, J.; Eterme, M. Control of the Immune Response by Pro-Angiogenic Factors. Front. Oncol. 2014, 4, 70. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Du Four, S.; Maenhout, S.K.; De Pierre, K.; Renmans, D.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology 2015, 4, e998107. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2012, 229, 176–185. [Google Scholar] [CrossRef]
- Komohara, Y.; Fujiwara, Y.; Ohnishi, K.; Takeya, M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016, 99, 180–185. [Google Scholar] [CrossRef]
- DiNapoli, M.R.; Calderon, C.L.; Lopez, D.M. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J. Exp. Med. 1996, 183, 1323–1329. [Google Scholar] [CrossRef]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.; Mantovani, A.; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, N.; Lederle, W.; Depner, S.; Van Rooijen, N.; Gutschalk, C.M.; Mueller, M.M. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J. Pathol. 2012, 227, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.J.; Pradeep, S.; McGuire, M.; Hailemichael, Y.; Ma, S.; Lyons, Y.; Armaiz-Pena, G.N.; Previs, R.A.; Hansen, J.M.; Rupaimoole, R.; et al. Macrophages Facilitate Resistance to Anti-VEGF Therapy by Altered VEGFR Expression. Clin. Cancer Res. 2017, 23, 7034–7046. [Google Scholar] [CrossRef] [Green Version]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Liu, D.; Cortes-Santiago, N.; Hossain, M.B.; Conrad, C.A.; Aldape, K.D.; Fuller, G.N.; Marini, F.C.; Alonso, M.M.; Idoate, M.A.; et al. Anti-vascular endothelial growth factor therapy-induced glioma invasion is associated with accumulation of Tie2-expressing monocytes. Oncotarget 2014, 5, 2208–2220. [Google Scholar] [CrossRef] [Green Version]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effectin vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018, 78, 4270–4281. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Zhou, J.; Dong, Z.; Tandon, S.; Kuk, D.; Panageas, K.S.; Wong, P.; Wu, X.; Naidoo, J.; Page, D.B.; et al. Pretreatment Serum VEGF Is Associated with Clinical Response and Overall Survival in Advanced Melanoma Patients Treated with Ipilimumab. Cancer Immunol. Res. 2014, 2, 127–132. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Connolly, E.M.; Liao, X.; Ouyang, J.; Giobbie-Hurder, A.; Lawrence, D.; McDermott, D.; Murphy, G.; Zhou, J.; et al. Combined Anti-VEGF and Anti–CTLA-4 Therapy Elicits Humoral Immunity to Galectin-1 Which Is Associated with Favorable Clinical Outcomes. Cancer Immunol. Res. 2017, 5, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D.; et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018, 19, 451–460. [Google Scholar] [CrossRef]
- Amin, A.; Dudek, A.Z.; Logan, T.F.; Lance, R.S.; Holzbeierlein, J.M.; Knox, J.J.; Master, V.A.; Pal, S.K.; Miller, W.H.; Karsh, L.I.; et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): Phase 2 study results. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, D.F.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.J.; Fong, L.; Joseph, R.W.; Pal, S.K.; Sznol, M.; Hainsworth, J.D.; et al. A phase II study of atezolizumab (atezo) with or without bevacizumab (bev) versus sunitinib (sun) in untreated metastatic renal cell carcinoma (mRCC) patients (pts). J. Clin. Oncol. 2017, 35, 431. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Ryoo, B.Y.; Hsu, C.H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Gagnon, M.L.; Bielenberg, D.R.; Gechtman, Z.; Miao, H.Q.; Takashima, S.; Soker, S.; Klagsbrun, M. Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: In vivo expression and antitumor activity. Proc. Natl. Acad. Sci. USA 2000, 97, 2573–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedam, S.B.; Low, J.A.; Yang, S.X.; Chow, C.K.; Choyke, P.; Danforth, D.; Hewitt, S.M.; Berman, A.; Steinberg, S.M.; Liewehr, D.J.; et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J. Clin. Oncol. 2006, 24, 769–777. [Google Scholar] [CrossRef] [PubMed]
- von Baumgarten, L.; Brucker, D.; Tirniceru, A.; Kienast, Y.; Grau, S.; Burgold, S.; Herms, J.; Winkler, F. Bevacizumab has differential and dose-dependent effects on glioma blood vessels and tumor cells. Clin. Cancer Res. 2011, 17, 6192–6205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, F.; Samuel, S.; Gaur, P.; Lu, J.; Dallas, N.A.; Xia, L.; Bose, D.; Ramachandran, V.; Ellis, L.M. Chronic exposure of colorectal cancer cells to bevacizumab promotes compensatory pathways that mediate tumour cell migration. Br. J. Cancer 2011, 104, 1270–1277. [Google Scholar] [CrossRef] [Green Version]
- Motz, G.T.; Coukos, G. The parallel lives of angiogenesis and immunosuppression: Cancer and other tales. Nat. Rev. Immunol. 2011, 11, 702–711. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]


{kind=link}
| Cancer Type | Effects |
|---|---|
| Melanoma (VEGFR1/2; NRP1/2) | Enhances the proliferation of melanoma cells [59]. Mitigates melanoma cells migration (through a NRP1/VEGFR2-mediated response) [60]. |
| Pancreatic (VEGFR1/2; NRP1) | Was shown to activate the MAPK/ERK pathway [44,61]. Stimulates cancer cell growth [44]. Promotes cancer cell migration and invasion, without affecting proliferation (VEGFR1-mediated effect) [61]. Promotes pancreatic cancer aggressiveness by TGFβ1-induced fibrosis and endothelial-to-mesenchymal transition (NRP1-mediated effect) [62]. |
| NSCLC (VEGFR1/2; NRP1/2) | Induces PI3K/Akt and MAPK/ERK activation [63]. Stimulates tumor growth and proliferation of NRP1-expressing cells (VEGFR2/NRP1-mediated effect) [63]. |
| SCLC (VEGFR2/3) | Promotes VEGFR2/3 activation resulting MAPK/ERK phosphorylation [64]. Induces cancer cell proliferation [64]. |
| Colorectal (VEGFR1) | Promotes Akt and ERK phosphorylation [65]. Enhances survival and resistance to chemotherapy of cancer cells [65]. Was shown to enhance cellular migration and promote tumor progression and metastasis [66,67]. Was found to support the survival of cancer cells undergoing EMT [68,69]. |
| Gastric (VEGFR1/2) | Stimulates tumor growth (VEGFR2-mediated response) [57,70]. |
| Prostate (VEGFR1/2) | Was shown to enhance prostate cancer cells proliferation (VEGFR2-mediated effect) [71,72]. |
| Glioblastoma (VEGFR1/2; NRP1) | Promotes MAPK/ERK, PI3K/Akt and PLC/PKC pathways activation [73,74]. Stimulates proliferation of glioma cells (VEGFR2-mediated response) [75]. Supports tumor growth (VEGFR1/2-mediated effect) [73]. |
| Breast cancer (VEGFR1/2; NRP1) | Induces activation of the MAPK/ERK and PI3K/Akt pathways [76]. Supports tumor cells survival, stimulates their proliferation and contributes to mammary tumor growth [77,78,79,80,81]. Induces invasion and chemotaxis of breast cancer cells and enhances EMT [79,80,81,82]. Inhibits apoptosis and protects from chemotherapy [79,83]. Confers cancer stem cells traits in breast cancer cells and was found to drive cancer stem cells self-renewal [84,85]. |
| Head & Neck (VEGFR2) | Regulates proliferation and invasion of head & neck cancer cells [86]. |
| Bladder (VEGFR1/2) | Enhances survival and proliferation of bladder cancer cells (VEGFR2-mediated effect) [45,87]. |
| Rhabdomyosarcoma (VEGFR1/2) | Increases cancer cell proliferation (VEGFR1-mediated effect) [52]. |
| Ovarian (VEGFR2) | VEGFR2-phosphorylation has been corelated with ovarian cancer cell survival and proliferation [58]. |
| Multiple Myeloma (VEGFR1) | Mediates activation of the MAPK/ERK, PI3 k/PKC and McL1/survivin pathways resulting in increased proliferation, migration and survival [88,89,90,91,92]. |
| Anti-Angiogenic Agent | Functions | |
|---|---|---|
| VEGF-A antibody | Bevacizumab | Decreases MDSCs and Tregs accumulation [124,125]. |
| Enhances CTLs responses: It was shown to (a) increases the peripheral B- and T-cell compartments [126], (b) correlate with an increase in activated (CD8+ CD62 L+) CTLs, long-term effector memory (CD8+ CD27+) and central-memory (CD8+ C45 RA-CCR7+) CTLs [127,128] and (c) enhance antigen-specific T-cell migration [129] | ||
| Improves DCs maturation and activation: It was shown to increase the percentage of activated and mature myeloid derived DC [127,130], and to reverse the VEGF inhibitory effects on DCs [131]. | ||
| Induces vessel normalization, increases tumor vascular expression of ICAM1 and VCAM1 and T-cell tumor infiltration [132,133,134,135]. | ||
| VEGFR1–3, PDGFR, c-KIT, FLT-3, CSF-1 R and RET mtTKI | Sunitinib | Enhances the Th1 immune response and inhibits the immunosuppressive Th2 response [136,137]. |
| Decreases MDSCs and tumor Tregs compartments [136,137,138,139,140]. | ||
| Induces endothelial activation and T-cell recruitment, by enhancing the expression of chemokines and adhesion molecules on tumor endothelial cells, resulting in a higher number of CD3+ T-cells in the tumor [141,142]. | ||
| Enhances the percentage and number of intratumoral CD4 and CD8 T-cells and decreases the expression of inhibitory molecules (i.e., CTLA-4 and PD-1) on TILs [141,143]. | ||
| VEGFR1–3, PDGFR and c-KIT mtTKI | Axitinib | Enhances the CD8+ T cells compartment [144]. |
| Increases the antigen-presenting function of intratumoral DCs [145]. | ||
| Reduces MDSCs levels [144] and inhibits their suppressive capacity [145]. | ||
| VEFGR2 TKI | Apatinib | Increases the infiltration of CD8+ T cells and reduces the recruitment of TAMs [146]. |
| Reduces the expression levels of inhibitory checkpoint molecules, such as Lag-3, PD-1 and Tim3 in CD8+ T cells [147]. | ||
| Enhances the production of IFN-γ and IL-2 and promote the cytotoxicity of T cells [147]. | ||
| Raf, VEGFR2, PDGFR, FLT3, RET and c-KIT mtTKI | Sorafenib | Reverses immunosuppression: It decreases MDSCs levels [148], Tregs and Th2-cells [149], and inhibits Tregs functions [150]. |
| Upregulates tumor-specific effector T-cells functions [150] and induces Th1 dominance [149]. | ||
| Reverses the VEGF inhibitory effects on DCs [131], but was also shown to inhibit the function of DCs [151] and inhibit the induction of antigen-specific T cells [151]. | ||
| Cancer Type | Immunotherapy | Anti-Angiogenic Agent | Indication | Year | Current Status | Identifier |
|---|---|---|---|---|---|---|
| Gastrointestinal | Atezolizumab (Anti-PDL1) | Bevacizumab | dMMR, Metastatic CRC | 2016 | Suspended | NCT02997228 |
| Nivolumab (Anti-PD1) | Bevacizumab | Metastatic CRC, 1st line | 2018 | Active, not recruiting | NCT03414983 | |
| Sintilimab (Anti-PD1) | Bevacizumab | RAS-Mutant, Metastatic CRC, 1st line | 2019 | Not yet recruiting | NCT04194359 | |
| HLX10 (Anti-PD1) | HLX04 (Anti-VEGF) | Metastatic CRC, 1st line | 2020 | Not yet recruiting | NCT04547166 | |
| Atezolizumab (Anti-PDL1) | Bevacizumab | Advanced HCC, 1st line | 2018 | Active, not recruiting | NCT03434379 | |
| HLX10 (Anti-PD1) | HLX04 (Anti-VEGF) | Advanced or Metastatic HCC, 1st line | 2020 | Not yet recruiting | NCT04465734 | |
| Genitourinary | Pembrolizumab (Anti-PD1) | Axitinib | Untreated, advanced RCC | 2016 | Active, not recruiting | NCT02853331 |
| Pembrolizumab (Anti-PD1) | Lenvatinib | Untreated, advanced RCC | 2016 | Active, not recruiting | NCT02811861 | |
| Atezolizumab (Anti-PDL1) | Bevacizumab | Untreated, advanced RCC | 2015 | Active, not recruiting | NCT02420821 | |
| Avelumab (Anti-PDL1) | Axitinib | Untreated, advanced RCC | 2016 | Active, not recruiting | NCT02684006 | |
| Nivolumab (Anti-PD1) | Cabozantinib | Untreated, metastatic RCC | 2019 | Recruiting | NCT03793166 | |
| Anlotinib (anti-PDL1) | TQB2450 (mtTKI) | Advanced RCC | 2020 | Recruiting | NCT04523272 | |
| Toripalimab (anti-PD1) | Axitinib | Unresectable or Metastatic RCC, 1st line | 2020 | Recruiting | NCT04394975 | |
| Lung | Atezolizumab (Anti-PDL1) | Bevacizumab | Stage IV Non-Squamous NSCLC, 1st line | 2015 | Active, not recruiting | NCT02366143 |
| Atezolizumab (Anti-PDL1) | Bevacizumab | Stage IV Non-Squamous NSCLC, 1st line | 2019 | Recruiting | NCT04194203 | |
| Sintilimab (Anti-PD1) | IBI305 (Anti-VEGF) | EGFR-mutated, TKI-resistant, Locally Advanced or Metastatic, non-squamous NSCLC | 2019 | Recruiting | NCT03802240 | |
| HLX10 (Anti-PD1) | HLX04 (Anti-VEGF) | Stage IIIB/IIIC or IV non-squamous NSCLC | 2019 | Recruiting | NCT03952403 | |
| Gynecological | Atezolizumab (Anti-PDL1) | Bevacizumab | Platinum-Resistant, Recurrent, Ovarian, Fallopian Tube, or Peritoneal Cancer | 2016 | Recruiting | NCT02839707 |
| Atezolizumab (Anti-PDL1) | Bevacizumab | Platinum-Sensitive Relapse, Ovarian, Fallopian Tube, or Peritoneal Cancer | 2016 | Active, not recruiting | NCT02891824 | |
| Atezolizumab (Anti-PDL1) | Bevacizumab | Stage III/IV Ovarian, Fallopian Tube, or Peritoneal Cancer | 2017 | Active, not recruiting | NCT03038100 | |
| Atezolizumab (Anti-PDL1) | Bevacizumab | Persistent, Recurrent or Metastatic (Stage IVB) Cervical Cancer | 2018 | Recruiting | NCT03556839 | |
| Pembrolizumab (Anti-PD1) | Bevacizumab | Persistent, Recurrent or Metastatic Cervical Cancer | 2018 | Active, not recruiting | NCT03635567 | |
| Dostarlimab (Anti-PD1) | Bevacizumab | Stage III/IV Nonmucinous Ovarian Cancer, 1st line | 2018 | Recruiting | NCT03602859 | |
| BCD-100 (Anti-PD1) | Bevacizumab | Advanced Cervical Cancer, 1st line | 2019 | Recruiting | NCT03912415 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ntellas, P.; Mavroeidis, L.; Gkoura, S.; Gazouli, I.; Amylidi, A.-L.; Papadaki, A.; Zarkavelis, G.; Mauri, D.; Karpathiou, G.; Kolettas, E.; et al. Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer. Cancers 2020, 12, 3145. https://doi.org/10.3390/cancers12113145
Ntellas P, Mavroeidis L, Gkoura S, Gazouli I, Amylidi A-L, Papadaki A, Zarkavelis G, Mauri D, Karpathiou G, Kolettas E, et al. Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer. Cancers. 2020; 12(11):3145. https://doi.org/10.3390/cancers12113145
Chicago/Turabian StyleNtellas, Panagiotis, Leonidas Mavroeidis, Stefania Gkoura, Ioanna Gazouli, Anna-Lea Amylidi, Alexandra Papadaki, George Zarkavelis, Davide Mauri, Georgia Karpathiou, Evangelos Kolettas, and et al. 2020. "Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer" Cancers 12, no. 11: 3145. https://doi.org/10.3390/cancers12113145
APA StyleNtellas, P., Mavroeidis, L., Gkoura, S., Gazouli, I., Amylidi, A. -L., Papadaki, A., Zarkavelis, G., Mauri, D., Karpathiou, G., Kolettas, E., Batistatou, A., & Pentheroudakis, G. (2020). Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer. Cancers, 12(11), 3145. https://doi.org/10.3390/cancers12113145