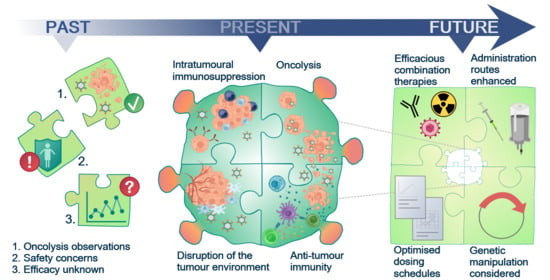
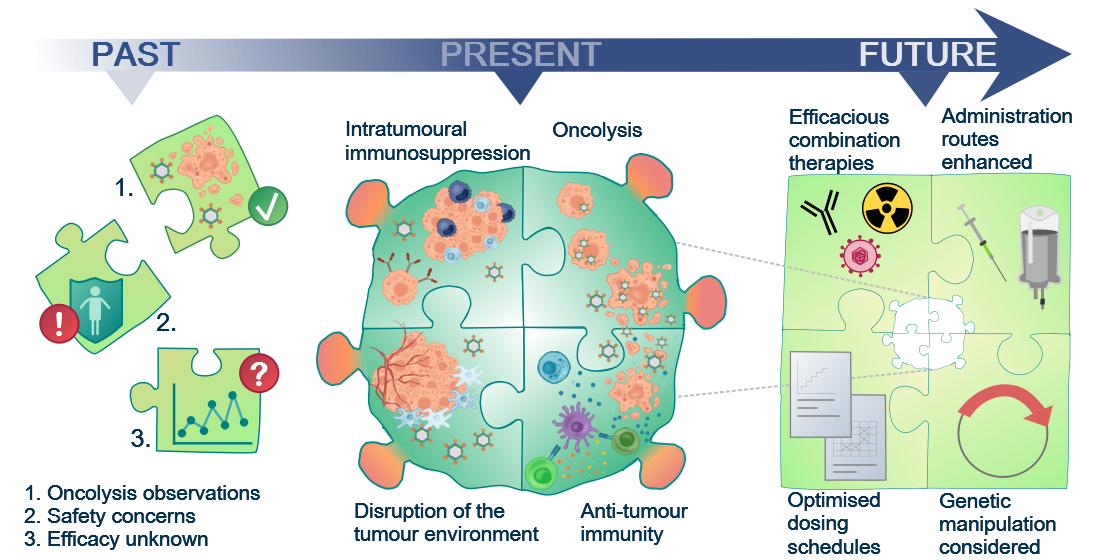
Past, Present and Future of Oncolytic Reovirus


Abstract
:Simple Summary
Abstract
1. Oncolytic Virotherapy (OVT)
2. The Emergence of Reovirus as a Therapeutic Agent
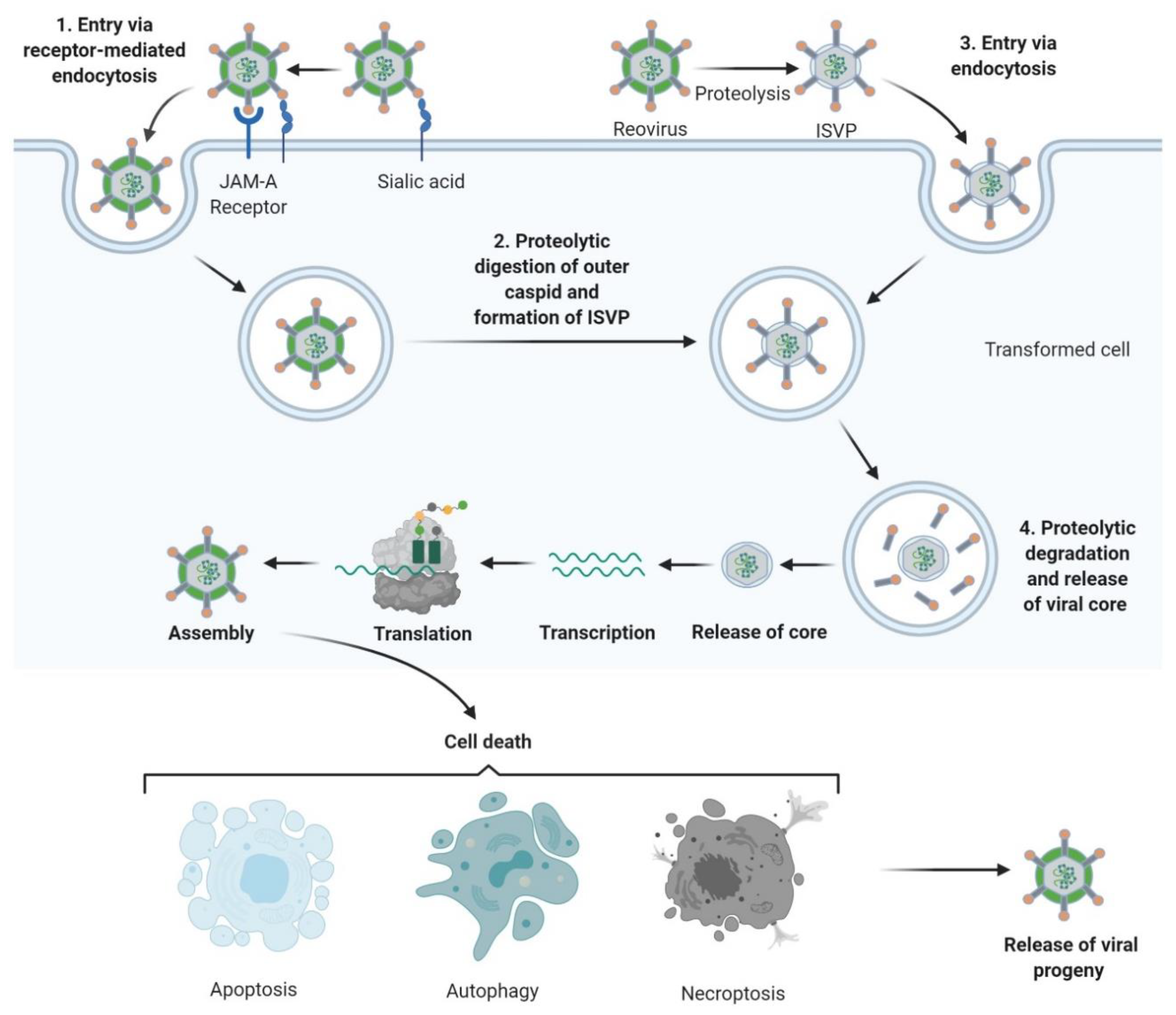
3. Tumour Specificity and Replication
4. Mechanisms of Oncolysis
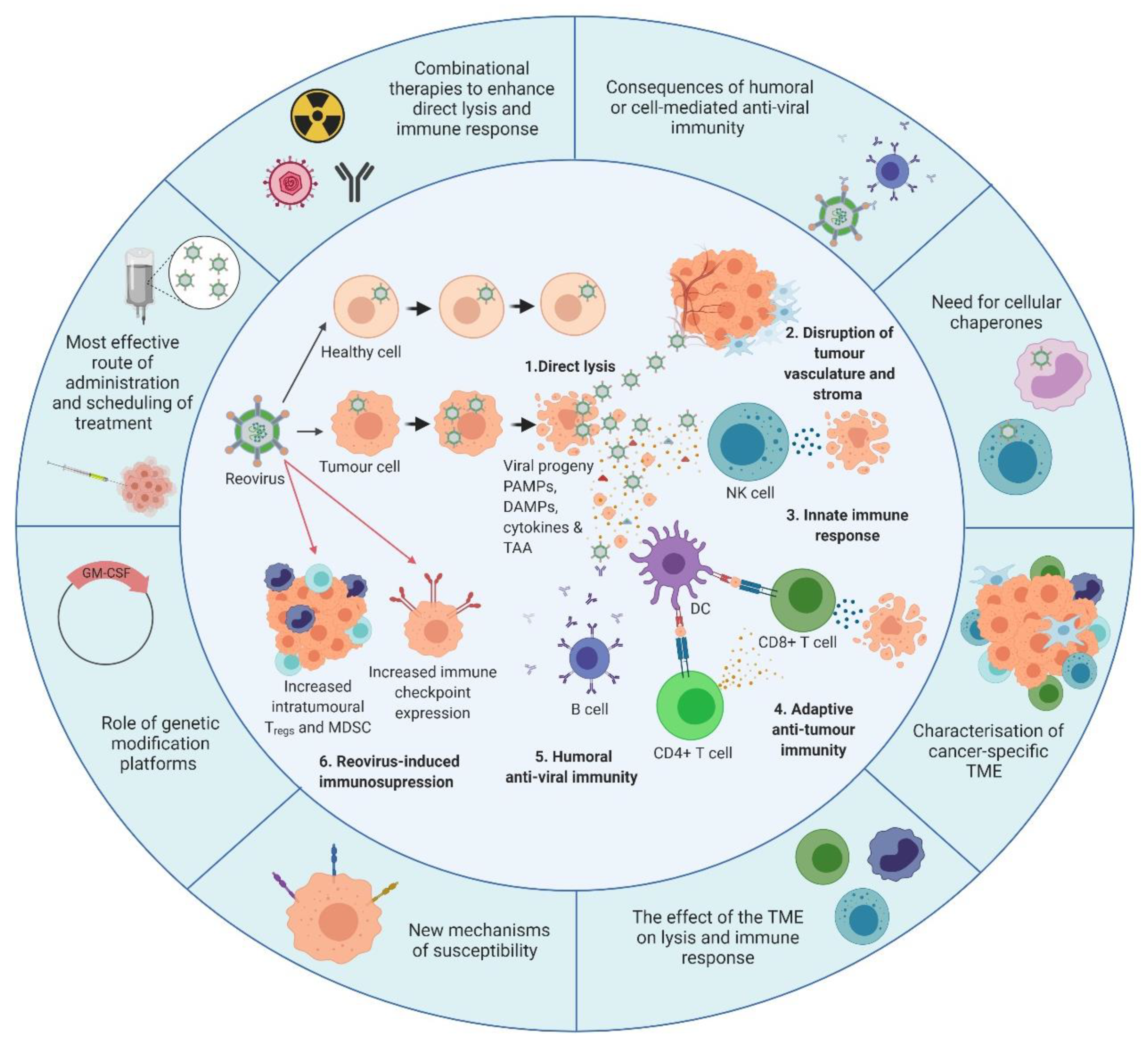
5. Reovirus Modulation of the Immune System
5.1. Reovirus-Induced Innate Anti-Tumour Immunity
5.2. Adaptive Anti-Tumour Immunity
5.3. The Antiviral Immune Response
6. Reovirus Delivery—Systemic vs. Intra-Tumoural
7. Unlocking the Potential of Reovirus with Combination Therapeutics
8. Reovirus Clinical Trials
9. The Future for Reovirus—Pre-Clinical Requirements and Clinical Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilkow, C.S.; Marguerie, M.; Batenchuk, C.; Mayer, J.; Ben Neriah, D.; Cousineau, S.; Falls, T.; Jennings, V.A.; Boileau, M.; Bellamy, D.; et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015, 21, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Bauzon, M.; Hermiston, T. Armed therapeutic viruses—A disruptive therapy on the horizon of cancer immunotherapy. Front. Immunol. 2014, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randazzo, B.P.; Tal-Singer, R.; Zabolotny, J.M.; Kesari, S.; Fraser, N.W. Herpes simplex virus 1716, an ICP 34.5 null mutant, is unable to replicate in CV-1 cells due to a translational block that can be overcome by coinfection with SV40. J. Gen. Virol 1997, 78 Pt 12, 3333–3339. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelli, D.; Beato, M.S.; Cavicchio, L.; Lavazza, A.; Chiapponi, C.; Leopardi, S.; Baioni, L.; De Benedictis, P.; Moreno, A. First identification of mammalian orthoreovirus type 3 in diarrheic pigs in Europe. Virol. J. 2016, 13, 139. [Google Scholar] [CrossRef] [Green Version]
- Steyer, A.; Gutierrez-Aguire, I.; Kolenc, M.; Koren, S.; Kutnjak, D.; Pokorn, M.; Poljsak-Prijatelj, M.; Racki, N.; Ravnikar, M.; Sagadin, M.; et al. High similarity of novel orthoreovirus detected in a child hospitalized with acute gastroenteritis to mammalian orthoreoviruses found in bats in Europe. J. Clin. Microbiol. 2013, 51, 3818–3825. [Google Scholar] [CrossRef] [Green Version]
- Rosen, L.; Hovis, J.F.; Mastrota, F.M.; Bell, J.A.; Huebner, R.J. Observations on a newly recognized virus (Abney) of the reovirus family. Am. J. Hyg. 1960, 71, 258–265. [Google Scholar] [CrossRef]
- Gaillard, R.K., Jr.; Joklik, W.K. Quantitation of the relatedness of reovirus serotypes 1, 2, and 3 at the gene level. Virology 1982, 123, 152–164. [Google Scholar] [CrossRef]
- Cashdollar, L.W.; Chmelo, R.A.; Wiener, J.R.; Joklik, W.K. Sequences of the S1 genes of the three serotypes of reovirus. Proc. Natl. Acad. Sci. USA 1985, 82, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiro, G.; Loh, P.C.; Yau, J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977, 54, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Alvarez, M.; Sabin, A.B. Characteristics of poliomyelitis and other enteric viruses recovered in tissue culture from healthy American children. Proc. Soc. Exp. Biol. Med. 1954, 87, 655–661. [Google Scholar] [CrossRef]
- Ramos-Alvarez, M.; Sabin, A.B. Enteropathogenic viruses and bacteria; role in summer diarrheal diseases of infancy and early childhood. J. Am. Med. Assoc. 1958, 167, 147–156. [Google Scholar] [CrossRef]
- Chua, K.B.; Crameri, G.; Hyatt, A.; Yu, M.; Tompang, M.R.; Rosli, J.; McEachern, J.; Crameri, S.; Kumarasamy, V.; Eaton, B.T.; et al. A previously unknown reovirus of bat origin is associated with an acute respiratory disease in humans. Proc. Natl. Acad. Sci. USA 2007, 104, 11424–11429. [Google Scholar] [CrossRef] [Green Version]
- Tillotson, J.R.; Lerner, A.M. Reovirus type 3 associated with fatal pneumonia. N. Engl. J. Med. 1967, 276, 1060–1063. [Google Scholar] [CrossRef]
- Ouattara, L.A.; Barin, F.; Barthez, M.A.; Bonnaud, B.; Roingeard, P.; Goudeau, A.; Castelnau, P.; Vernet, G.; Paranhos-Baccala, G.; Komurian-Pradel, F. Novel human reovirus isolated from children with acute necrotizing encephalopathy. Emerg. Infect. Dis. 2011, 17, 1436–1444. [Google Scholar] [CrossRef]
- Tyler, K.L.; Barton, E.S.; Ibach, M.L.; Robinson, C.; Campbell, J.A.; O’Donnell, S.M.; Valyi-Nagy, T.; Clarke, P.; Wetzel, J.D.; Dermody, T.S. Isolation and molecular characterization of a novel type 3 reovirus from a child with meningitis. J. Infect. Dis. 2004, 189, 1664–1675. [Google Scholar] [CrossRef] [Green Version]
- Morecki, R.; Glaser, J.H.; Cho, S.; Balistreri, W.F.; Horwitz, M.S. Biliary atresia and reovirus type 3 infection. N. Engl. J. Med. 1982, 307, 481–484. [Google Scholar] [CrossRef]
- Richardson, S.C.; Bishop, R.F.; Smith, A.L. Reovirus serotype 3 infection in infants with extrahepatic biliary atresia or neonatal hepatitis. J. Gastroenterol. Hepatol. 1994, 9, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Dermody, T.; Parker, J.; Sherry, B. Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1304–1346. [Google Scholar]
- Lemay, G. Transcriptional and translational events during reovirus infection. Biochem. Cell Biol. 1988, 66, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Shatkin, A.J.; Sipe, J.D.; Loh, P. Separation of ten reovirus genome segments by polyacrylamide gel electrophoresis. J. Virol. 1968, 2, 986–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, K.; Zhang, X.; Olson, N.H.; Walker, S.B.; Chappell, J.D.; Dermody, T.S.; Baker, T.S.; Nibert, M.L. Complete in vitro assembly of the reovirus outer capsid produces highly infectious particles suitable for genetic studies of the receptor-binding protein. J. Virol. 2001, 75, 5335–5342. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Shatkin, A.J. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology 1997, 234, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanifer, M.L.; Kischnick, C.; Rippert, A.; Albrecht, D.; Boulant, S. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci. Rep. 2017, 7, 10873. [Google Scholar] [CrossRef]
- Barton, E.S.; Connolly, J.L.; Forrest, J.C.; Chappell, J.D.; Dermody, T.S. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 2001, 276, 2200–2211. [Google Scholar] [CrossRef] [Green Version]
- Chappell, J.D.; Duong, J.L.; Wright, B.W.; Dermody, T.S. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 2000, 74, 8472–8479. [Google Scholar] [CrossRef] [Green Version]
- Antar, A.A.; Konopka, J.L.; Campbell, J.A.; Henry, R.A.; Perdigoto, A.L.; Carter, B.D.; Pozzi, A.; Abel, T.W.; Dermody, T.S. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 2009, 5, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Barton, E.S.; Forrest, J.C.; Connolly, J.L.; Chappell, J.D.; Liu, Y.; Schnell, F.J.; Nusrat, A.; Parkos, C.A.; Dermody, T.S. Junction adhesion molecule is a receptor for reovirus. Cell 2001, 104, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Shmulevitz, M.; Gujar, S.A.; Ahn, D.G.; Mohamed, A.; Lee, P.W. Reovirus variants with mutations in genome segments S1 and L2 exhibit enhanced virion infectivity and superior oncolysis. J. Virol. 2012, 86, 7403–7413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugano, Y.; Takeuchi, M.; Hirata, A.; Matsushita, H.; Kitamura, T.; Tanaka, M.; Miyajima, A. Junctional adhesion molecule-A, JAM-A, is a novel cell-surface marker for long-term repopulating hematopoietic stem cells. Blood 2008, 111, 1167–1172. [Google Scholar] [CrossRef]
- Parrish, C.; Scott, G.B.; Migneco, G.; Scott, K.J.; Steele, L.P.; Ilett, E.; West, E.J.; Hall, K.; Selby, P.J.; Buchanan, D.; et al. Oncolytic reovirus enhances rituximab-mediated antibody-dependent cellular cytotoxicity against chronic lymphocytic leukaemia. Leukemia 2015, 29, 1799–1810. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275–41289. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Scott, K.J.; Rose, A.; Desborough, M.; Harrington, K.; Pandha, H.; Parrish, C.; Vile, R.; Coffey, M.; Bowen, D.; et al. Reovirus-mediated cytotoxicity and enhancement of innate immune responses against acute myeloid leukemia. BioRes. Open Access 2012, 1, 3–15. [Google Scholar] [CrossRef]
- Xu, P.P.; Sun, Y.F.; Fang, Y.; Song, Q.; Yan, Z.X.; Chen, Y.; Jiang, X.F.; Fei, X.C.; Zhao, Y.; Leboeuf, C.; et al. JAM-A overexpression is related to disease progression in diffuse large B-cell lymphoma and downregulated by lenalidomide. Sci. Rep. 2017, 7, 7433. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lu, F.; Chen, H.; Zhao, X.; Sun, J.; Chen, H. Dysregulation of JAM-A plays an important role in human tumor progression. Int. J. Clin. Exp. Pathol. 2014, 7, 7242–7248. [Google Scholar]
- Zhang, M.; Luo, W.; Huang, B.; Liu, Z.; Sun, L.; Zhang, Q.; Qiu, X.; Xu, K.; Wang, E. Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS ONE 2013, 8, e79173. [Google Scholar] [CrossRef] [Green Version]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef]
- Alain, T.; Kim, T.S.; Lun, X.; Liacini, A.; Schiff, L.A.; Senger, D.L.; Forsyth, P.A. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1512–1521. [Google Scholar] [CrossRef]
- Golden, J.W.; Linke, J.; Schmechel, S.; Thoemke, K.; Schiff, L.A. Addition of exogenous protease facilitates reovirus infection in many restrictive cells. J. Virol. 2002, 76, 7430–7443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, D.H.; Deussing, J.; Peters, C.; Dermody, T.S. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002, 277, 24609–24617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, K.; Farsetta, D.L.; Nibert, M.L. Strategy for nonenveloped virus entry: A hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J. Virol. 2002, 76, 9920–9933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibert, M.L.; Odegard, A.L.; Agosto, M.A.; Chandran, K.; Schiff, L.A. Putative autocleavage of reovirus mu1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 2005, 345, 461–474. [Google Scholar] [CrossRef]
- Odegard, A.L.; Chandran, K.; Zhang, X.; Parker, J.S.; Baker, T.S.; Nibert, M.L. Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus. J. Virol. 2004, 78, 8732–8745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.K.; Scheible, P.P.; Keene, J.D.; Joklik, W.K. The plus strand of reovirus gene S2 is identical with its in vitro transcript. Virology 1980, 105, 282–286. [Google Scholar] [CrossRef]
- Fields, B.N.; Raine, C.S.; Baum, S.G. Temperature-sensitive mutants of reovirus type 3: Defects in viral maturation as studied by immunofluorescence and electron microscopy. Virology 1971, 43, 569–578. [Google Scholar] [CrossRef]
- Kobayashi, T.; Chappell, J.D.; Danthi, P.; Dermody, T.S. Gene-specific inhibition of reovirus replication by RNA interference. J. Virol. 2006, 80, 9053–9063. [Google Scholar] [CrossRef] [Green Version]
- Antczak, J.B.; Joklik, W.K. Reovirus genome segment assortment into progeny genomes studied by the use of monoclonal antibodies directed against reovirus proteins. Virology 1992, 187, 760–776. [Google Scholar] [CrossRef]
- McDonald, S.M.; Patton, J.T. Assortment and packaging of the segmented rotavirus genome. Trends Microbiol. 2011, 19, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, J.L.; Barton, E.S.; Dermody, T.S. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 2001, 75, 4029–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.M.; Mainou, B.A.; Kim, K.S.; Dermody, T.S. Directional release of reovirus from the apical surface of polarized endothelial cells. mBio 2013, 4, e00049-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, J.E.; Tang, D.; Lee, P.W. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology 1993, 197, 405–411. [Google Scholar] [CrossRef]
- Tang, D.; Strong, J.E.; Lee, P.W. Recognition of the epidermal growth factor receptor by reovirus. Virology 1993, 197, 412–414. [Google Scholar] [CrossRef]
- Strong, J.E.; Lee, P.W. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J. Virol. 1996, 70, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Shmulevitz, M.; Marcato, P.; Lee, P.W. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene 2005, 24, 7720–7728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, K.L.; Hirasawa, K.; Yang, A.D.; Shields, M.A.; Lee, P.W. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef] [Green Version]
- Smakman, N.; van den Wollenberg, D.J.; Borel Rinkes, I.H.; Hoeben, R.C.; Kranenburg, O. Sensitization to apoptosis underlies KrasD12-dependent oncolysis of murine C26 colorectal carcinoma cells by reovirus T3D. J. Virol. 2005, 79, 14981–14985. [Google Scholar] [CrossRef] [Green Version]
- Sadler, A.J.; Williams, B.R. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 2007, 316, 253–292. [Google Scholar] [CrossRef]
- Christian, S.L.; Zu, D.; Licursi, M.; Komatsu, Y.; Pongnopparat, T.; Codner, D.A.; Hirasawa, K. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS ONE 2012, 7, e44267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Haro, C.; Mendez, R.; Santoyo, J. The eIF-2alpha kinases and the control of protein synthesis. FASEB J. 1996, 10, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Mita, M.M. Activated ras signaling pathways and reovirus oncolysis: An update on the mechanism of preferential reovirus replication in cancer cells. Front. Oncol. 2014, 4, 167. [Google Scholar] [CrossRef]
- Kim, M.; Egan, C.; Alain, T.; Urbanski, S.J.; Lee, P.W.; Forsyth, P.A.; Johnston, R.N. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene 2007, 26, 4124–4134. [Google Scholar] [CrossRef]
- Smakman, N.; van der Bilt, J.D.; van den Wollenberg, D.J.; Hoeben, R.C.; Borel Rinkes, I.H.; Kranenburg, O. Immunosuppression promotes reovirus therapy of colorectal liver metastases. Cancer Gene Ther. 2006, 13, 815–818. [Google Scholar] [CrossRef]
- Song, L.; Ohnuma, T.; Gelman, I.H.; Holland, J.F. Reovirus infection of cancer cells is not due to activated Ras pathway. Cancer Gene Ther. 2009, 16, 382. [Google Scholar] [CrossRef] [Green Version]
- Twigger, K.; Roulstone, V.; Kyula, J.; Karapanagiotou, E.M.; Syrigos, K.N.; Morgan, R.; White, C.; Bhide, S.; Nuovo, G.; Coffey, M.; et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer 2012, 12, 368. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.; Richardson-Burns, S.M.; DeBiasi, R.L.; Tyler, K.L. Mechanisms of apoptosis during reovirus infection. Curr. Top. Microbiol. Immunol. 2005, 289, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, A.; Haque, S.J.; Mogensen, T.; Silverman, R.H.; Williams, B.R. RNA-dependent protein kinase PKR is required for activation of NF-kappa B by IFN-gamma in a STAT1-independent pathway. J. Immunol. 2001, 166, 6170–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Sherry, B. Rotavirus and reovirus modulation of the interferon response. J. Interferon Cytokine Res. 2009, 29, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Schmechel, S.C.; Williams, B.R.; Silverman, R.H.; Schiff, L.A. Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J. Virol. 2005, 79, 2240–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, P.; Meintzer, S.M.; Gibson, S.; Widmann, C.; Garrington, T.P.; Johnson, G.L.; Tyler, K.L. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 2000, 74, 8135–8139. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, D.J.; Bickel, R.J.; Tyler, K.L. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 2002, 9, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, D.J.; Bickel, R.J.; Tyler, K.L. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 2002, 76, 11414–11424. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, J.J.; Dermody, T.S.; Holm, G.H. Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-kappaB-dependent expression of Noxa. J. Virol. 2012, 86, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.K.; Danthi, P. Reovirus activates a caspase-independent cell death pathway. mBio 2013, 4, e00178-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E., Jr.; Upton, J.W.; Danthi, P. Viral RNA at Two Stages of Reovirus Infection Is Required for the Induction of Necroptosis. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 2013, 9, 413–414. [Google Scholar] [CrossRef] [Green Version]
- Errington, F.; Steele, L.; Prestwich, R.; Harrington, K.J.; Pandha, H.S.; Vidal, L.; de Bono, J.; Selby, P.; Coffey, M.; Vile, R.; et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J. Immunol. 2008, 180, 6018–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douville, R.N.; Su, R.C.; Coombs, K.M.; Simons, F.E.; Hayglass, K.T. Reovirus serotypes elicit distinctive patterns of recall immunity in humans. J. Virol. 2008, 82, 7515–7523. [Google Scholar] [CrossRef] [Green Version]
- Errington, F.; White, C.L.; Twigger, K.R.; Rose, A.; Scott, K.; Steele, L.; Ilett, L.J.; Prestwich, R.; Pandha, H.S.; Coffey, M.; et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008, 15, 1257–1270. [Google Scholar] [CrossRef]
- Steele, L.; Errington, F.; Prestwich, R.; Ilett, E.; Harrington, K.; Pandha, H.; Coffey, M.; Selby, P.; Vile, R.; Melcher, A. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol. Cancer 2011, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Wetzel, J.D.; He, J.; Mikacenic, C.; Dermody, T.S.; Kelsall, B.L. Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J. Exp. Med. 2007, 204, 1349–1358. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Marie, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, M.; Arias, C.F.; Lopez, S. Protein kinase R is responsible for the phosphorylation of eIF2alpha in rotavirus infection. J. Virol. 2010, 84, 10457–10466. [Google Scholar] [CrossRef] [Green Version]
- Adair, R.A.; Scott, K.J.; Fraser, S.; Errington-Mais, F.; Pandha, H.; Coffey, M.; Selby, P.; Cook, G.P.; Vile, R.; Harrington, K.J.; et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int. J. Cancer 2013, 132, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Pan, L.Z.; Garant, K.; Pan, D.; Lee, P.W. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010, 70, 4912–4921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, Y.; Tachibana, M.; Kurisu, N.; Oya, Y.; Terasawa, Y.; Goda, H.; Kobiyama, K.; Ishii, K.J.; Akira, S.; Mizuguchi, H.; et al. Oncolytic Reovirus Inhibits Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in a TLR3-Dependent Manner. J. Immunol 2018, 200, 2987–2999. [Google Scholar] [CrossRef]
- Cai, J.; Lin, Y.; Zhang, H.; Liang, J.; Tan, Y.; Cavenee, W.K.; Yan, G. Selective replication of oncolytic virus M1 results in a bystander killing effect that is potentiated by Smac mimetics. Proc. Natl. Acad. Sci. USA 2017, 114, 6812–6817. [Google Scholar] [CrossRef] [Green Version]
- Arulanandam, R.; Batenchuk, C.; Varette, O.; Zakaria, C.; Garcia, V.; Forbes, N.E.; Davis, C.; Krishnan, R.; Karmacharya, R.; Cox, J.; et al. Microtubule disruption synergizes with oncolytic virotherapy by inhibiting interferon translation and potentiating bystander killing. Nat. Commun. 2015, 6, 6410. [Google Scholar] [CrossRef] [Green Version]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef] [Green Version]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Mohamed-Hadley, A.; Zhang, L.; Buckanovich, R.J.; Carroll, R.; Fraser, N.; Coukos, G. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 789–802. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Errington, F.; Steele, L.P.; Ilett, E.J.; Morgan, R.S.; Harrington, K.J.; Pandha, H.S.; Selby, P.J.; Vile, R.G.; Melcher, A.A. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J. Immunol 2009, 183, 4312–4321. [Google Scholar] [CrossRef] [Green Version]
- Spiotto, M.T.; Rowley, D.A.; Schreiber, H. Bystander elimination of antigen loss variants in established tumors. Nat. Med. 2004, 10, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Ilett, E.J.; Morgan, R.S.; Scott, K.J.; Kottke, T.; Thompson, J.; Morrison, E.E.; Harrington, K.J.; Pandha, H.S.; et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin. Cancer Res. 2008, 14, 7358–7366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, S.A.; Pan, D.A.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic virus-initiated protective immunity against prostate cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.T.; Roy, D.G.; Workenhe, S.T.; van den Wollenberg, D.J.M.; Hoeben, R.C.; Mossman, K.L.; Bell, J.C.; Bourgeois-Daigneault, M.C. Pre-surgical neoadjuvant oncolytic virotherapy confers protection against rechallenge in a murine model of breast cancer. Sci. Rep. 2019, 9, 1865. [Google Scholar] [CrossRef] [Green Version]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Clements, D.R.; Sterea, A.M.; Kim, Y.; Helson, E.; Dean, C.A.; Nunokawa, A.; Coyle, K.M.; Sharif, T.; Marcato, P.; Gujar, S.A.; et al. Newly recruited CD11b+, GR-1+, Ly6C(high) myeloid cells augment tumor-associated immunosuppression immediately following the therapeutic administration of oncolytic reovirus. J. Immunol. 2015, 194, 4397–4412. [Google Scholar] [CrossRef] [Green Version]
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized Phase 2 Trial of the Oncolytic Virus Pelareorep (Reolysin) in Upfront Treatment of Metastatic Pancreatic Adenocarcinoma. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef] [Green Version]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wu, K.; Visconte, V.; Anwer, F.; Calton, C.M.; Carew, J.S.; Nawrocki, S.T. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia 2018, 32, 230–233. [Google Scholar] [CrossRef]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [Green Version]
- White, C.L.; Twigger, K.R.; Vidal, L.; De Bono, J.S.; Coffey, M.; Heinemann, L.; Morgan, R.; Merrick, A.; Errington, F.; Vile, R.G.; et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008, 15, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sherbiny, Y.M.; Holmes, T.D.; Wetherill, L.F.; Black, E.V.; Wilson, E.B.; Phillips, S.L.; Scott, G.B.; Adair, R.A.; Dave, R.; Scott, K.J.; et al. Controlled infection with a therapeutic virus defines the activation kinetics of human natural killer cells in vivo. Clin. Exp. Immunol. 2015, 180, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Wang, H.; Kottke, T.; White, C.; Twigger, K.; Diaz, R.M.; Thompson, J.; Selby, P.; de Bono, J.; Melcher, A.; et al. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin. Cancer Res. 2008, 14, 259–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, K.; Ishikura, M.; Nakayama, T.; Hasegawa, S.; Morita, O.; Uetake, H. Ecological studies on reovirus pollution of rivers in Toyama Prefecture. Microbiol. Immunol. 1988, 32, 1221–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minuk, G.Y.; Paul, R.W.; Lee, P.W. The prevalence of antibodies to reovirus type 3 in adults with idiopathic cholestatic liver disease. J. Med. Virol. 1985, 16, 55–60. [Google Scholar] [CrossRef]
- Lerner, A.M.; Cherry, J.D.; Klein, J.O.; Finland, M. Infections with reoviruses. N. Engl. J. Med. 1962, 267, 947–952. [Google Scholar] [CrossRef]
- Selb, B.; Weber, B. A study of human reovirus IgG and IgA antibodies by ELISA and western blot. J. Virol. Methods 1994, 47, 15–25. [Google Scholar] [CrossRef]
- Pal, S.R.; Agarwal, S.C. Sero-epidemiological study of reovirus infection amongst the normal population of the Chandigarh area—Northern India. J. Hyg. (Lond.) 1968, 66, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Tai, J.H.; Williams, J.V.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr.; Dermody, T.S. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J. Infect. Dis. 2005, 191, 1221–1224. [Google Scholar] [CrossRef]
- Li, X.; Wang, P.; Li, H.; Du, X.; Liu, M.; Huang, Q.; Wang, Y.; Wang, S. The Efficacy of Oncolytic Adenovirus Is Mediated by T-cell Responses against Virus and Tumor in Syrian Hamster Model. Clin. Cancer Res. 2017, 23, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Alain, T.; Kossakowska, A.; Lee, P.W. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002, 62, 1696–1701. [Google Scholar]
- Norman, K.L.; Coffey, M.C.; Hirasawa, K.; Demetrick, D.J.; Nishikawa, S.G.; DiFrancesco, L.M.; Strong, J.E.; Lee, P.W. Reovirus oncolysis of human breast cancer. Hum. Gene Ther. 2002, 13, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Sei, S.; Mussio, J.K.; Yang, Q.E.; Nagashima, K.; Parchment, R.E.; Coffey, M.C.; Shoemaker, R.H.; Tomaszewski, J.E. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol. Cancer 2009, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Thirukkumaran, C.M.; Nodwell, M.J.; Hirasawa, K.; Shi, Z.Q.; Diaz, R.; Luider, J.; Johnston, R.N.; Forsyth, P.A.; Magliocco, A.M.; Lee, P.; et al. Oncolytic viral therapy for prostate cancer: Efficacy of reovirus as a biological therapeutic. Cancer Res. 2010, 70, 2435–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin. Cancer Res. 2012, 18, 4962–4972. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef]
- Qiao, J.; Kottke, T.; Willmon, C.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Ryno, P.; Barber, G.N.; Chester, J.; et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.E.; Yang, W.; Senger, D.; Rewcastle, N.B.; Morris, D.G.; Brasher, P.M.; Shi, Z.Q.; Johnston, R.N.; Nishikawa, S.; Lee, P.W.; et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J. Natl. Cancer Inst. 2001, 93, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Samson, A.; Bentham, M.J.; Scott, K.; Nuovo, G.; Bloy, A.; Appleton, E.; Adair, R.A.; Dave, R.; Peckham-Cooper, A.; Toogood, G.; et al. Oncolytic reovirus as a combined antiviral and anti-tumour agent for the treatment of liver cancer. Gut 2018, 67, 562–573. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Adair, R.A.; Roulstone, V.; Scott, K.J.; Morgan, R.; Nuovo, G.J.; Fuller, M.; Beirne, D.; West, E.J.; Jennings, V.A.; Rose, A.; et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 2012, 4, 138ra77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scurr, M.; Pembroke, T.; Bloom, A.; Roberts, D.; Thomson, A.; Smart, K.; Bridgeman, H.; Adams, R.; Brewster, A.; Jones, R.; et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 6771–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurd, E.R.; Giuliano, V.J. The effect of cyclophosphamide on B and T lymphocytes in patients with connective tissue diseases. Arthritis Rheum 1975, 18, 67–75. [Google Scholar] [CrossRef]
- Varkila, K.; Hurme, M. The effect of cyclophosphamide on cytotoxic T-lymphocyte responses: Inhibition of helper T-cell induction in vitro. Immunology 1983, 48, 433–438. [Google Scholar]
- Ikeda, K.; Ichikawa, T.; Wakimoto, H.; Silver, J.S.; Deisboeck, T.S.; Finkelstein, D.; Harsh, G.R.t.; Louis, D.N.; Bartus, R.T.; Hochberg, F.H.; et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat. Med. 1999, 5, 881–887. [Google Scholar] [CrossRef]
- Peng, K.W.; Myers, R.; Greenslade, A.; Mader, E.; Greiner, S.; Federspiel, M.J.; Dispenzieri, A.; Russell, S.J. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013, 20, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.; Ball, M.; Tanay, M.; Nutting, C.; Newbold, K.; Gore, M.E.; Larkin, J.; Syrigos, K.N.; et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012, 18, 2080–2089. [Google Scholar] [CrossRef] [Green Version]
- Lolkema, M.P.; Arkenau, H.T.; Harrington, K.; Roxburgh, P.; Morrison, R.; Roulstone, V.; Twigger, K.; Coffey, M.; Mettinger, K.; Gill, G.; et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2011, 17, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Roulstone, V.; Khan, K.; Pandha, H.S.; Rudman, S.; Coffey, M.; Gill, G.M.; Melcher, A.A.; Vile, R.; Harrington, K.J.; de Bono, J.; et al. Phase I trial of cyclophosphamide as an immune modulator for optimizing oncolytic reovirus delivery to solid tumors. Clin. Cancer Res. 2015, 21, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Markovic, S.N.; Suman, V.J.; Nuovo, G.J.; Vile, R.G.; Kottke, T.J.; Nevala, W.K.; Thompson, M.A.; Lewis, J.E.; Rumilla, K.M.; et al. Phase II trial of intravenous administration of Reolysin((R)) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1998–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.; Scurr, M.; Campbell, E.; Jones, E.; Godkin, A.; Gallimore, A. T-cell modulation by cyclophosphamide for tumour therapy. Immunology 2018, 154, 62–68. [Google Scholar] [CrossRef]
- Huang, X.M.; Zhang, N.R.; Lin, X.T.; Zhu, C.Y.; Zou, Y.F.; Wu, X.J.; He, X.S.; He, X.W.; Wan, Y.L.; Lan, P. Antitumor immunity of low-dose cyclophosphamide: Changes in T cells and cytokines TGF-beta and IL-10 in mice with colon-cancer liver metastasis. Gastroenterol. Rep. (Oxf.) 2020, 8, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Ilett, E.J.; Barcena, M.; Errington-Mais, F.; Griffin, S.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Limpens, R.W.; Mommaas, M.; et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin. Cancer Res. 2011, 17, 2767–2776. [Google Scholar] [CrossRef] [Green Version]
- Ilett, E.; Kottke, T.; Donnelly, O.; Thompson, J.; Willmon, C.; Diaz, R.; Zaidi, S.; Coffey, M.; Selby, P.; Harrington, K.; et al. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Berkeley, R.A.; Steele, L.P.; Mulder, A.A.; van den Wollenberg, D.J.M.; Kottke, T.J.; Thompson, J.; Coffey, M.; Hoeben, R.C.; Vile, R.G.; Melcher, A.; et al. Antibody-Neutralized Reovirus Is Effective in Oncolytic Virotherapy. Cancer Immunol. Res. 2018, 6, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Jennings, V.A.; Ilett, E.J.; Scott, K.J.; West, E.J.; Vile, R.; Pandha, H.; Harrington, K.; Young, A.; Hall, G.D.; Coffey, M.; et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int. J. Cancer 2014, 134, 1091–1101. [Google Scholar] [CrossRef]
- Twigger, K.; Vidal, L.; White, C.L.; De Bono, J.S.; Bhide, S.; Coffey, M.; Thompson, B.; Vile, R.G.; Heinemann, L.; Pandha, H.S.; et al. Enhanced in vitro and in vivo cytotoxicity of combined reovirus and radiotherapy. Clin. Cancer Res. 2008, 14, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, P.; Zhang, W.; Lin, J.; Liu, L.; Guha, C.; Kolb, E.A. Systemic administration of reovirus (Reolysin) inhibits growth of human sarcoma xenografts. Cancer 2011, 117, 1764–1774. [Google Scholar] [CrossRef]
- Heinemann, L.; Simpson, G.R.; Boxall, A.; Kottke, T.; Relph, K.L.; Vile, R.; Melcher, A.; Prestwich, R.; Harrington, K.J.; Morgan, R.; et al. Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer 2011, 11, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandha, H.S.; Heinemann, L.; Simpson, G.R.; Melcher, A.; Prestwich, R.; Errington, F.; Coffey, M.; Harrington, K.J.; Morgan, R. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin. Cancer Res. 2009, 15, 6158–6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulstone, V.; Twigger, K.; Zaidi, S.; Pencavel, T.; Kyula, J.N.; White, C.; McLaughlin, M.; Seth, R.; Karapanagiotou, E.M.; Mansfield, D.; et al. Synergistic cytotoxicity of oncolytic reovirus in combination with cisplatin-paclitaxel doublet chemotherapy. Gene Ther. 2013, 20, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Gujar, S.A.; Clements, D.; Dielschneider, R.; Helson, E.; Marcato, P.; Lee, P.W. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br. J. Cancer 2014, 110, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death: Where the cell cycle and apoptosis come together. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Safarian, S.; Carroll, J.S.; Sheibani, N.; Bidkhori, G. Tamoxifen-Induced Apoptosis of MCF-7 Cells via GPR30/PI3K/MAPKs Interactions: Verification by ODE Modeling and RNA Sequencing. Front. Physiol. 2018, 9, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, R.; Rabb, M.; Madureira, P.A.; Clements, D.; Gujar, S.A.; Waisman, D.M.; Giacomantonio, C.A.; Lee, P.W. Gemcitabine-mediated tumour regression and p53-dependent gene expression: Implications for colon and pancreatic cancer therapy. Cell Death Dis. 2013, 4, e791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, D.L.; Gray, H.; Payne, C.M.; Gillies, R.J. Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells. Mol. Cancer Ther. 2005, 4, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Espitia, C.M.; Mahalingam, D.; Oyajobi, B.O.; Coffey, M.; Giles, F.J.; Carew, J.S.; Nawrocki, S.T. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene 2012, 31, 3023–3038. [Google Scholar] [CrossRef] [Green Version]
- Roulstone, V.; Pedersen, M.; Kyula, J.; Mansfield, D.; Khan, A.A.; McEntee, G.; Wilkinson, M.; Karapanagiotou, E.; Coffey, M.; Marais, R.; et al. BRAF- and MEK-Targeted Small Molecule Inhibitors Exert Enhanced Antimelanoma Effects in Combination With Oncolytic Reovirus Through ER Stress. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.E.; Sadek, M.; Gujar, S.A. Targeted Metabolic Reprogramming to Improve the Efficacy of Oncolytic Virus Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.E.; Murphy, J.P.; Clements, D.R.; Konda, P.; Holay, N.; Kim, Y.; Pathak, G.P.; Giacomantonio, M.A.; Hiani, Y.E.; Gujar, S. Inhibition of Pyruvate Dehydrogenase Kinase Enhances the Antitumor Efficacy of Oncolytic Reovirus. Cancer Res. 2019, 79, 3824–3836. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Hall, G.; Pulido, J.; Diaz, R.M.; Thompson, J.; Chong, H.; Selby, P.; Coffey, M.; Pandha, H.; Chester, J.; et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J. Clin. Investig. 2010, 120, 1551–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottke, T.; Chester, J.; Ilett, E.; Thompson, J.; Diaz, R.; Coffey, M.; Selby, P.; Nuovo, G.; Pulido, J.; Mukhopadhyay, D.; et al. Precise scheduling of chemotherapy primes VEGF-producing tumors for successful systemic oncolytic virotherapy. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1802–1812. [Google Scholar] [CrossRef] [Green Version]
- Ilett, E.; Kottke, T.; Thompson, J.; Rajani, K.; Zaidi, S.; Evgin, L.; Coffey, M.; Ralph, C.; Diaz, R.; Pandha, H.; et al. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumour therapy. Gene Ther. 2017, 24, 21–30. [Google Scholar] [CrossRef]
- Mostafa, A.A.; Meyers, D.E.; Thirukkumaran, C.M.; Liu, P.J.; Gratton, K.; Spurrell, J.; Shi, Q.; Thakur, S.; Morris, D.G. Oncolytic Reovirus and Immune Checkpoint Inhibition as a Novel Immunotherapeutic Strategy for Breast Cancer. Cancers (Basel) 2018, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.G.; Feng, X.; DiFrancesco, L.M.; Fonseca, K.; Forsyth, P.A.; Paterson, A.H.; Coffey, M.C.; Thompson, B. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin(R)) in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 696–706. [Google Scholar] [CrossRef]
- Gollamudi, R.; Ghalib, M.H.; Desai, K.K.; Chaudhary, I.; Wong, B.; Einstein, M.; Coffey, M.; Gill, G.M.; Mettinger, K.; Mariadason, J.M.; et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Investig. New Drugs 2010, 28, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Vidal, L.; Pandha, H.S.; Yap, T.A.; White, C.L.; Twigger, K.; Vile, R.G.; Melcher, A.; Coffey, M.; Harrington, K.J.; DeBono, J.S. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin. Cancer Res. 2008, 14, 7127–7137. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, P.; Roldan, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef]
- Sborov, D.W.; Nuovo, G.J.; Stiff, A.; Mace, T.; Lesinski, G.B.; Benson, D.M., Jr.; Efebera, Y.A.; Rosko, A.E.; Pichiorri, F.; Grever, M.R.; et al. A phase I trial of single-agent reolysin in patients with relapsed multiple myeloma. Clin. Cancer Res. 2014, 20, 5946–5955. [Google Scholar] [CrossRef] [Green Version]
- Karnad, A.B.; Haigentz, M.; Miley, T.; Coffey, M.; Gill, G.; Mita, M. Abstract C22: A phase II study of intravenous wild-type reovirus (Reolysin®) in combination with paclitaxel plus carboplatin in patients with platinum refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. Mol. Cancer Ther. 2011, 10. [Google Scholar] [CrossRef]
- Ocean, A.J.; Bekaii-Saab, T.S.; Chaudhary, I.; Palmer, R.; Christos, P.J.; Mercado, A.; Florendo, E.O.; Rosales, V.A.; Ruggiero, J.T.; Popa, E.C.; et al. A multicenter phase I study of intravenous administration of reolysin in combination with irinotecan/fluorouracil/leucovorin (FOLFIRI) in patients (pts) with oxaliplatin-refractory/intolerant KRAS-mutant metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2013, 31, 450. [Google Scholar] [CrossRef]
- Villalona-Calero, M.A.; Lam, E.; Otterson, G.A.; Zhao, W.; Timmons, M.; Subramaniam, D.; Hade, E.M.; Gill, G.M.; Coffey, M.; Selvaggi, G.; et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer 2016, 122, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, V.E.; Dent, S.F.; Gelmon, K.A.; Dhesy-Thind, S.K.; Mates, M.; Salim, M.; Panasci, L.; Song, X.; Clemons, M.; Tu, D.; et al. A randomized (RCT) phase II study of oncolytic reovirus (pelareorep ) plus standard weekly paclitaxel (P) as therapy for metastatic breast cancer (mBC) [Abstract nr CT131]. In Proceedings of the American Association for Cancer Research Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef]
- Mita, A.; Sankhala, K.; Sarantopoulos, J. A phase II study of intravenous (IV) wild-type reovirus (Reolysin) in the treatment of patients with bone and soft tissue sarcomas metastatic to the lung. J. Clin. Oncol. 2009, 27, 10524. [Google Scholar] [CrossRef]
- Mahalingam, D.; Fountzilas, C.; Moseley, J.L.; Noronha, N.; Cheetham, K.; Dzugalo, A.; Nuovo, G.; Gutierrez, A.; Arora, S.P. A study of REOLYSIN in combination with pembrolizumab and chemotherapy in patients (pts) with relapsed metastatic adenocarcinoma of the pancreas (MAP). J. Clin. Oncol. 2017, 35, e15753. [Google Scholar] [CrossRef]
- Saunders, M.; Anthoney, A.; Coffey, M.; Mettinger, K.; Thompson, B.; Melcher, A.; Nutting, C.M.; Harrington, K. Results of a phase II study to evaluate the biological effects of intratumoral (ITu) reolysin in combination with low dose radiotherapy (RT) in patients (Pts) with advanced cancers. J. Clin. Oncol. 2009, 27. [Google Scholar] [CrossRef]
- Jonker, D.J.; Tang, P.A.; Kennecke, H.; Welch, S.A.; Cripps, M.C.; Asmis, T.; Chalchal, H.; Tomiak, A.; Lim, H.; Ko, Y.J.; et al. A Randomized Phase II Study of FOLFOX6/Bevacizumab With or Without Pelareorep in Patients With Metastatic Colorectal Cancer: IND.210, a Canadian Cancer Trials Group Trial. Clin. Colorectal. Cancer 2018, 17, 231–239.e7. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Fountzilas, C.; Moseley, J.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Coffey, M.; Thompson, B.; Sarantopoulos, J. A phase II study of REOLYSIN((R)) (pelareorep) in combination with carboplatin and paclitaxel for patients with advanced malignant melanoma. Cancer Chemother. Pharmcol. 2017, 79, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Mita, A.C.; Argiris, A.; Coffey, M.; Gill, G.; Mita, M. Abstract C70: A phase 2 study of intravenous administration of REOLYSIN® (reovirus type 3 dearing) in combination with paclitaxel (P) and carboplatin (C) in patients with squamous cell carcinoma of the lung. Mol. Cancer Ther. 2013, 12. [Google Scholar] [CrossRef]
- Bradbury, P.A.; Morris, D.G.; Nicholas, G.; Tu, D.; Tehfe, M.; Goffin, J.R.; Shepherd, F.A.; Gregg, R.W.; Rothenstein, J.; Lee, C.; et al. Canadian Cancer Trials Group (CCTG) IND211: A randomized trial of pelareorep (Reolysin) in patients with previously treated advanced or metastatic non-small cell lung cancer receiving standard salvage therapy. Lung Cancer (Amst. Neth.) 2018, 120, 142–148. [Google Scholar] [CrossRef]
- Eigl, B.J.; Chi, K.; Tu, D.; Hotte, S.J.; Winquist, E.; Booth, C.M.; Canil, C.; Potvin, K.; Gregg, R.; North, S.; et al. A randomized phase II study of pelareorep and docetaxel or docetaxel alone in men with metastatic castration resistant prostate cancer: CCTG study IND 209. Oncotarget 2018, 9, 8155–8164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, D.E.; Sill, M.W.; Walker, J.L.; O’Malley, D.; Nagel, C.I.; Rutledge, T.L.; Bradley, W.; Richardson, D.L.; Moxley, K.M.; Aghajanian, C. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin(R)) in recurrent ovarian, tubal, or peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 146, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.R.; White, C.L.; Vidal, L.; Beirne, D.; Prestwich, R.; Newbold, K.; Ahmed, M.; et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin. Cancer Res. 2010, 16, 3067–3077. [Google Scholar] [CrossRef] [Green Version]
- Comins, C.; Spicer, J.; Protheroe, A.; Roulstone, V.; Twigger, K.; White, C.M.; Vile, R.; Melcher, A.; Coffey, M.C.; Mettinger, K.L.; et al. REO-10: A phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin. Cancer Res. 2010, 16, 5564–5572. [Google Scholar] [CrossRef] [Green Version]
- van den Wollenberg, D.J.; Dautzenberg, I.J.; van den Hengel, S.K.; Cramer, S.J.; de Groot, R.J.; Hoeben, R.C. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS ONE 2012, 7, e48064. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.; Johnston, R.N.; Shmulevitz, M. Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants. Viruses 2015, 7, 6251–6278. [Google Scholar] [CrossRef] [Green Version]
- Kemp, V.; Hoeben, R.C.; van den Wollenberg, D.J. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2016, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipinska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene 2015, 22, 267–279. [Google Scholar] [CrossRef]
- Kemp, V.; van den Wollenberg, D.J.M. Arming oncolytic reovirus with GM-CSF gene to enhance immunity. Cancer Gene Ther. 2019, 26, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Stewart, R.M.; Berry, J.T.L.; Berger, A.K.; Yoon, S.B.; Hirsch, A.L.; Guberman, J.A.; Patel, N.B.; Tharp, G.K.; Bosinger, S.E.; Mainou, B.A. Enhanced Killing of Triple-Negative Breast Cancer Cells by Reassortant Reovirus and Topoisomerase Inhibitors. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Fernandes, J.P.; Cristi, F.; Eaton, H.E.; Chen, P.; Haeflinger, S.; Bernard, I.; Hitt, M.M.; Shmulevitz, M. Breast Tumor-Associated Metalloproteases Restrict Reovirus Oncolysis by Cleaving the σ1 Cell Attachment Protein and Can Be Overcome by Mutation of σ1. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Disease | Combinations | Phase | Trial ID | Route | Dose(s) TCID50 | Results |
|---|---|---|---|---|---|---|
| Gliomas | N/A | I | NCT00528684 | I.T | 1 × 107, 1 × 108, 1 × 109 | No DLT, 10/12 patients had PD, 1/12 SD and 1/12 patients unevaluable for response, but alive >4.5 years post treatment [171]. |
| N/A | I | EudraCT 2011-005635-10 | I.V | 1 × 1010 | Reovirus detected in within tumours and increased CTL infiltration [109]. | |
| Brain cancer | Sargramostim (GM-CSF) | I | NCT02444546 | I.V | MTD | Ongoing |
| Pancreatic cancer | Carboplatin and Paclitaxel | II | NCT01280058 | I.V | 3 × 1010 | No significant enhancement of PFS with reovirus combination therapy (n = 36) vs. Carboplatin/Paclitaxel alone (n = 37) (4.9 vs. 5.2 months) [108]. |
| Pembrolizumab and 5 Fluorouracil or gemcitabine or irinotecan | I | NCT02620423 | I.V | 4.5 × 1010 | Well tolerated. 3/10 evaluable patients had SD, 1 of which had PR for 17.4 months. Biopsies show reovirus infection in tumour cells and immune infiltrates [179]. | |
| Pembrolizumab | II | NCT03723915 | I.V | Not reported | Ongoing | |
| Gemcitabine | II | NCT00998322 | I.V | 1 × 1010 | Well tolerated. 1/29 patients had PR, 23/29 SD, 5/29 PD. Single patient with SD had upregulated expression of PD-L1 following treatment [180]. | |
| Colorectal cancer | Irinotecan and Leucovorin and 5-Fluorouracil | I | NCT01274624 | I.V | 1 × 1010– 3 × 1010 | 2/21 patients had DLT. 18/21 evaluable for response. 1/18 PR, 9/18 SD, 8/18 PD [174]. |
| Leucovorin and 5-Fluorouracil and Oxaliplatin and Bevacizumab | II | NCT01622543 | I.V | 3 × 1010 | Poorer PFS with reovirus combination therapy (7 months vs. 9 months). No significant difference in OS [181]. | |
| Head and Neck Cancers | Carboplatin and Paclitaxel | II | NCT00753038 | I.V | 3 × 1010 | Well tolerated. 4/13 evaluable patients had PR, 2/13 had SD for >12 weeks [173]. |
| Carboplatin and Paclitaxel | III | NCT01166542 | I.V | 3 × 1010 | Interim results reported (www.oncolyticsbiotech.com). 118 evaluable patients, reovirus increased PFS from 48 to 95 days. Significantly increased OS. Curtailed to larger phase II trial. | |
| Melanoma | N/A | II | NCT00651157 | I.V | 3 × 1010 | Well tolerated, viral replication was detected in 2/15 patients despite NAb, average PFS 45 days [142]. |
| Carboplatin and Paclitaxel | II | NCT00984464 | I.V | 3 × 1010 | Well tolerated. 3/14 patients had PR, 9/14 SD, 2/14 PD. ORR of 21%, no complete responses [182]. | |
| Multiple Myeloma | N/A | I | NCT01533194 | I.V | 3 × 109, 3 × 1010 | No DLT reported, reovirus localization to BM, SD for up to 8 months [172]. |
| Lenalidomide or Pomalidomide | I | NCT03015922 | I.V | 3 × 1010 | Ongoing | |
| Dexamethasone and Carfilzomib | I | NCT02101944 | I.V | MTD | Recruiting | |
| Dexamethasone and Bortezomib | I | NCT02514382 | I.V | MTD up to 4.5 × 1010 | Ongoing | |
| Dexamethasone and Carfilzomib and Nivolumab | I | NCT03605719 | I.V | MTD | Recruiting | |
| Lung Cancer | Carboplatin or Paclitaxel | II | NCT00861627 | I.V | 3 × 1010 | 11/37 of patients PR, 20/37 SD, PFS 4 months [175]. |
| II | NCT00998192 | I.V | 3 × 1010 | Treatment well tolerated, 12/25 patients had PR, 10/25 SD, 3/25 PD [183]. | ||
| Pemetrexed or Docetaxel | II | NCT01708993 | I.V | 4.5 × 1010 | Virus was well tolerated, no enhancement of PFS with reovirus vs. drugs alone (2.96 vs. 2.83 months) [184]. | |
| Prostate cancer | Docetaxel and Prednisone | II | NCT01619813 | I.V | 3 × 1010 | Poorer OS in virus and drug combination arm, vs. drug alone [185]. |
| Breast cancer | Paclitaxel | II | NCT01656538 | I.V | 3 × 1010 | Combination arm showed improved OS vs. drug alone arm (17.4 vs. 10.4 months) [176]. |
| Avelumab and Paclitaxel | II | NCT04215146 | I.V | 4.5 × 1010 | Recruiting | |
| Retifanlimab | II | NCT04445844 | I.V | MTD | Recruiting | |
| Ovarian cancer | Paclitaxel | II | NCT01166542 | I.V | 3 × 1010 | Median PFS 4.3 months and ORR 20% for patients receiving Pacitaxel alone vs. 4.4 months and 17.4%, for combination treatment. Addition of reovirus to treatment does not reduce the hazard of progression or death [186]. |
| Bone and soft tissue sarcoma | N/A | II | NCT00503295 | I.V | 3 × 1010 | Well tolerated. 14/33 patients had SD for >2 months, including 5 patients which had SD for >6 months [178]. |
| Advanced cancer | Radiotherapy | I | I.T | 1 × 108–1 × 1010 | No DLT. Low dose radiation arm 2/7 PR and 5/7 SD. High dose radiation arm 5/7 PR and 2/7 SD [187]. | |
| Carboplatin and Paclitaxel | I | I.V | 3 × 109, 1 × 1010, 3 × 1010 | No DLT. 1/26 patients had CR, 6/26 PR, 9/26 SD, 2/25 major clinical response, and 9/25 PD [139]. | ||
| Docetaxel | I | I.V | 3 × 109, 1 × 1010, 3 × 1010 | MTD not reached. 1/16 patients had CR, 3/16 PR, 3/16 minor response, 7/16 SD, 2/16 PD [188]. | ||
| Gemcitabine | I | I.V | 1 × 109 3 × 109, 1 × 1010, 3 × 1010 | 3/16 patients had DLT. 10/16 patients evaluable for response, 1/10 PR, 6/10 SD, 3/10 PD [140]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, L.; Berkeley, R.; Barr, T.; Ilett, E.; Errington-Mais, F. Past, Present and Future of Oncolytic Reovirus. Cancers 2020, 12, 3219. https://doi.org/10.3390/cancers12113219
Müller L, Berkeley R, Barr T, Ilett E, Errington-Mais F. Past, Present and Future of Oncolytic Reovirus. Cancers. 2020; 12(11):3219. https://doi.org/10.3390/cancers12113219
Chicago/Turabian StyleMüller, Louise, Robert Berkeley, Tyler Barr, Elizabeth Ilett, and Fiona Errington-Mais. 2020. "Past, Present and Future of Oncolytic Reovirus" Cancers 12, no. 11: 3219. https://doi.org/10.3390/cancers12113219
APA StyleMüller, L., Berkeley, R., Barr, T., Ilett, E., & Errington-Mais, F. (2020). Past, Present and Future of Oncolytic Reovirus. Cancers, 12(11), 3219. https://doi.org/10.3390/cancers12113219