Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment



Abstract
:Simple Summary
Abstract
1. Introduction
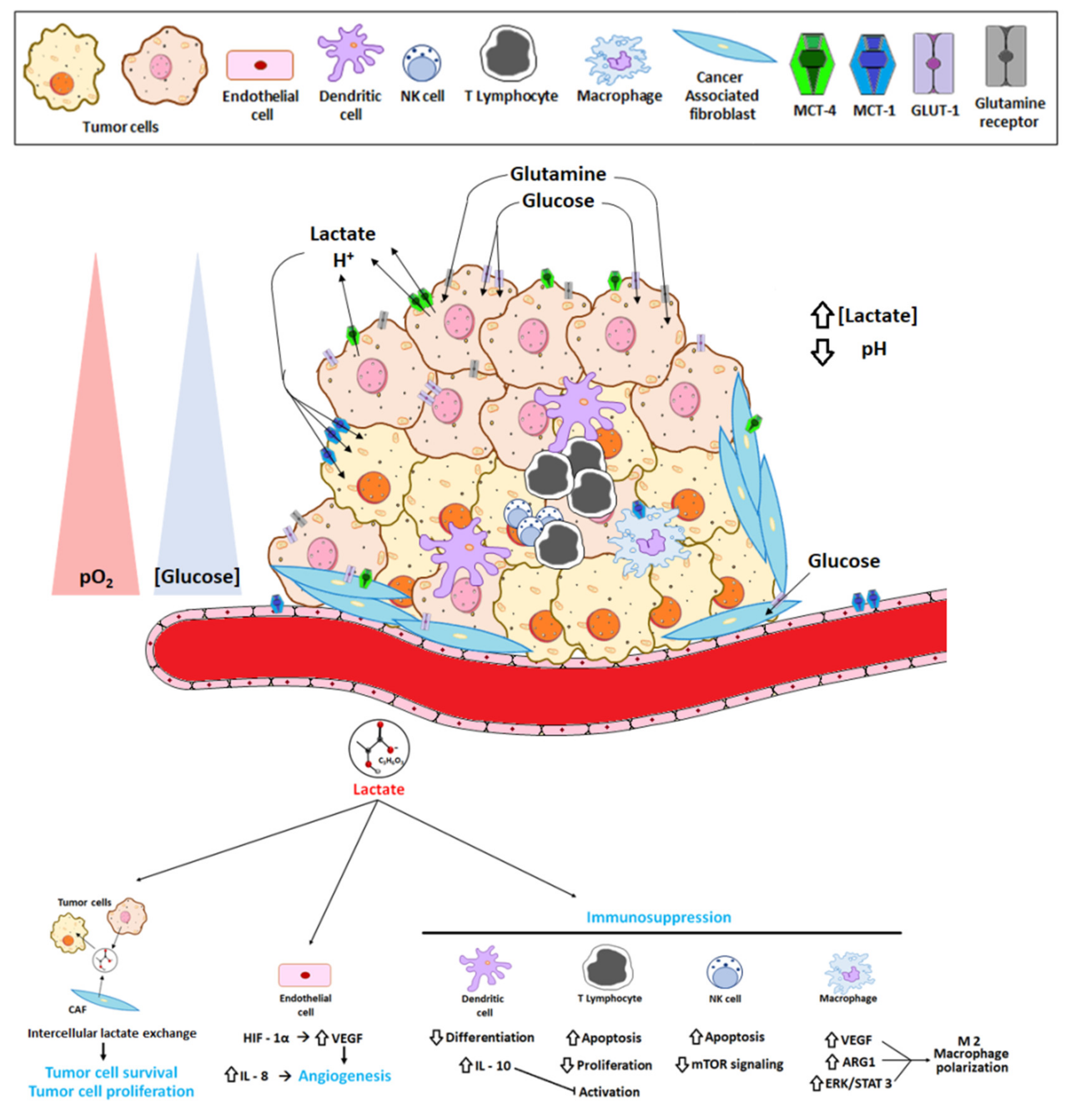
2. The Tumor Microenvironment and the Reversed pH Gradient
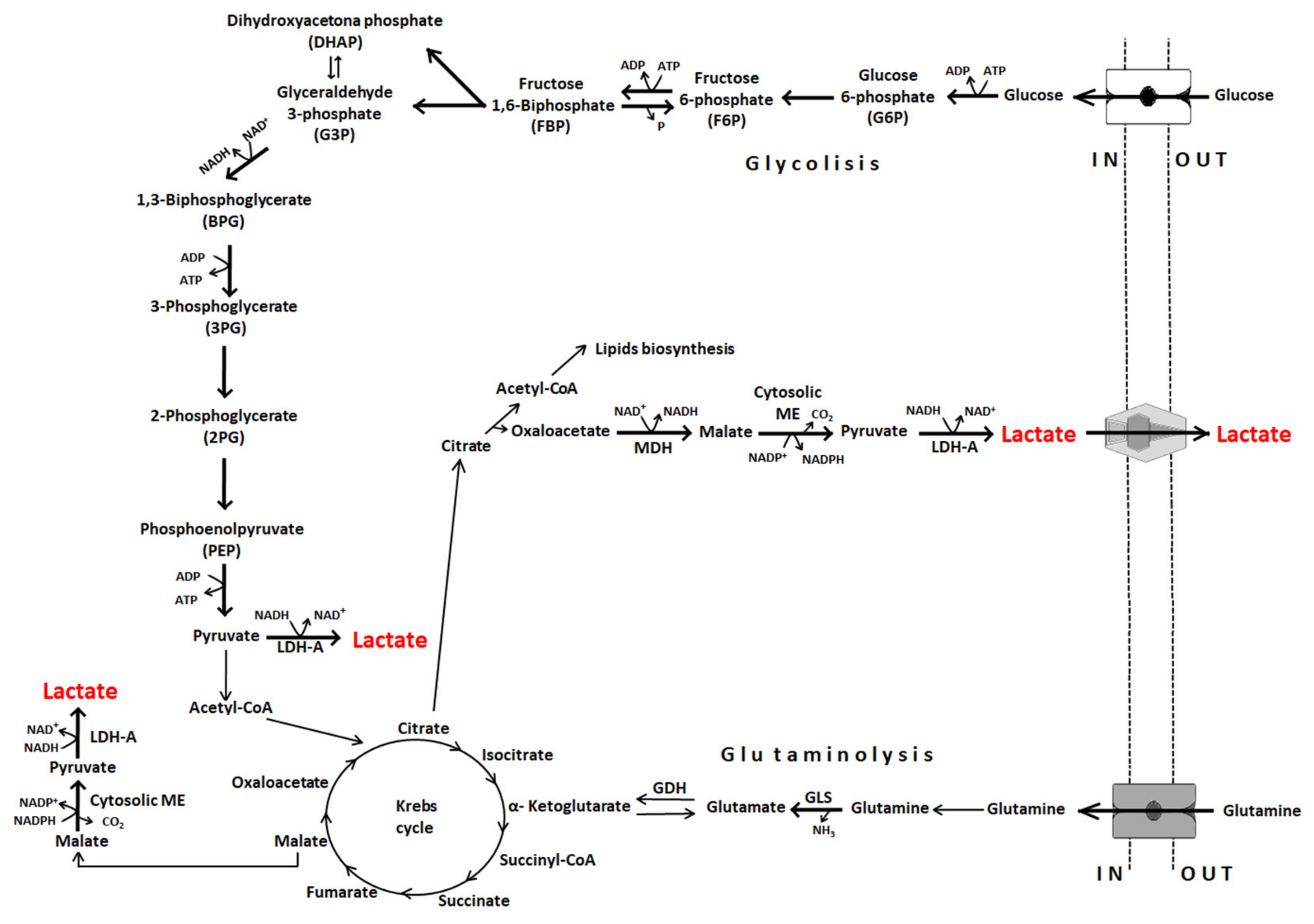
3. Lactate Dehydrogenases
4. Lactate Transport
5. The Lactate Receptor GPR81 and other Modes of Lactate Transport
6. Lactate Exchange
7. Lactate and Cancer-Related Genes
8. Lactate as a Key Molecule in the “Immune Scape”
9. Lactate in Tumor Metastasis
10. Lactate in Therapy Resistance
11. Therapeutic Strategies Targeting Lactate
11.1. Targeting Lactate Production
11.2. Targeting Lactate Transporters
12. Conclusions and Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Raju, T.N. The Nobel Chronicles. 1922: Archilbald Vivian Hill (1886-1977), Otto Fritz Meyerfhoff (1884-1951). Lancet 1998, 352, 1396. [Google Scholar] [CrossRef]
- Shampo, M.A.; Kyle, R.A. Otto Meyerhoff--Nobel Prize for studies of muscle metabolism. Mayo Clin. Proc. 1999, 74, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, A.; Macdonald, A.L.; Watt, P.W. Lactate-A signal coordinating cell and systemic function. J. Exp. Biol. 2005, 208, 4561–4575. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Damiani, C.; Colombo, R.; Gaglio, D.; Mastroianni, F.; Pescini, D.; Westerhoff, H.V.; Mauri, G.; Vanoni, M.; Alberghina, L. A metabolic core model elucidates how enhanced utilization of glucose and glutamine, with enhanced glutamine-dependent lactate production, promotes cancer cell growth: The Warburg effect. PLoS Comput. Biol. 2017, 13, e1005758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef] [Green Version]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, L.; Esposito, F.; Thomson, T.M.; Maurel, J. The Tumor Microenvironment in Colorectal Cancer Therapy. Cancers 2019, 11, 1172. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar] [PubMed]
- Brizel, D.M.; Schroeder, T.; Scher, R.L.; Walenta, S.; Clough, R.W.; Dewhirst, M.W.; Mueller-Klieser, W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 349–353. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Tatum, J.L.; Kelloff, G.J.; Gillies, R.J.; Arbeit, J.M.; Brown, J.M.; Chao, K.S.; Chapman, J.D.; Eckelman, W.C.; Fyles, A.W.; Giaccia, A.J. Hypoxia: Importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int. J. Radiat. Oncol. Biol. 2006, 82, 699–757. [Google Scholar] [CrossRef]
- Hashim, A.I.; Zhang, X.; Wojtkowiak, J.W.; Martínez, G.V.; Gillies, R.J. Imaging pH and metastasis. NMR Biomed. 2011, 24, 582–591. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Greco, M.R.; Cardone, R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130100. [Google Scholar] [CrossRef] [Green Version]
- Oberhaensli, R.D.; Bore, P.J.; Rampling, R.P.; Hilton-Jones, D.; Hands, L.J.; Radda, G.K. Biochemical investigation of human tumors in vivo with phosphorus-31 magnetic resonance spectroscopy. Lancet 1986, 328, 8–11. [Google Scholar] [CrossRef]
- Parks, S.K.; Chicha, J.; Pouysségur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef]
- Nuccitelli, R.; Deamer, D.W. Intracellular pH: Its measurement, regulation and utilization in cellular functions. Yale J. Biol Med. 1982, 55, 541–542. [Google Scholar]
- Andrés, V.; Carreras, J.; Cussó, R. Regulation of muscle phosphofructokinase by physiological concentrations of bisphosphorylated hexoses: Effect of alkalinization. Biochem. Biophys. Res. Commun. 1990, 172, 328–334. [Google Scholar] [CrossRef]
- Kuwata, F.; Suzuki, N.; Otsuka, K.; Taguchi, M.; Sasai, Y.; Wakino, H.; Ito, M.; Ebihara, S.; Suzuki, K. Enzymatic regulation of glycolysis and gluconeogenesis in rabbit periodontal ligament under various physiological pH conditions. J. Nihon Univ. Sch. Dent. 1991, 33, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.F. The Na + /H+ exchanger NHE1 in stress-induced signal transduction: Implications for cell proliferation and cell death. Pflug. Arch. 2006, 452, 249–259. [Google Scholar] [CrossRef]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, J.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Redd, J.C. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2000, 2, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Robergs, R.A.; Ghiasvand, F.; Parker, D. Biochemistry of the exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R502–R516. [Google Scholar] [CrossRef]
- Stubbs, M.; Rodrigues, L.; Howe, F.A.; Wang, J.; Jeong, K.S.; Veech, R.L.; Griffiths, J.R. Metabolic consequences of a reversed pH gradient in rat tumors. Cancer Res. 1994, 54, 4011–4016. [Google Scholar]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The regulation and function of lactate dehydrogenase A: Therapeutic potential in brain tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Gao, J.; Zhuang, X.; Zhao, C.; Hou, X.; Xing, X.; Chen, C.; Liu, Q.; Liu, S.; Luo, Y. Cyclin G2 inhibits the warburg effect and tumour progression by suppressing LDHA phosphorylation in glioma. Int. J. Biol. Sci. 2019, 15, 544–555. [Google Scholar] [CrossRef] [Green Version]
- Ždralević, M.; Marchiq, I.; De Padua, M.M.C.; Parks, S.K.; Pouysségur, J. Metabolic plasticity in cancers–distinct role of glycolytic enzymes GPI, LDHs or membrane transporters MCTs. Front. Oncol. 2017, 7, 313. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, X.; Ye, F.; Chen, B.; Song, C.; Wen, J.; Zhang, Z.; Zheng, G.; Tang, H.; Xie, X. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Sci. Rep. 2016, 6, 21735. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, Y.; Yan, X.; Song, Q.; Wang, G.; Hu, Y.; Jiao, S.; Wang, J. Pretreatment lactate dehydrogenase may predict outcome of advanced non-small-cell lung cancer patients treated with immune checkpoint inhibitors: A meta-analysis. Cancer Med. 2019, 8, 1467–1473. [Google Scholar] [CrossRef]
- Lv, J.; Zhou, Z.; Wang, J.; Yu, H.; Lu, H.; Yuan, B.; Han, J.; Zhou, R.; Zhang, X.; Yang, X.; et al. Prognostic Value of Lactate Dehydrogenase Expression in Different Cancers: A Meta-Analysis. Am. J. Med. Sci. 2019, 358, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Dawson, D.M.; Goodfriend, T.L.; Kaplan, N.O. Lactic dehydrogenases: Functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science 2013, 143, 929–933. [Google Scholar] [CrossRef]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.-J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer cells. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Zhang, P.; Wang, B.; Yang, D.; Duan, X.; Jiang, Y.; Xu, T.; Jiang, Y.; Shi, J.; Ding, C.; et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat. Commun. 2019, 10, 5566. [Google Scholar] [CrossRef] [Green Version]
- Urbańska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 2085. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, B.; Danforth, W.H. Effect of pH on the kinetics of frog muscle phosphofructokinase. J. Biol. Chem. 1966, 241, 4110–4112. [Google Scholar]
- Prado-Garcia, H.; Campa-Higareda, A.; Romero-Garcia, S. Lactic Acidosis in the Presence of Glucose Diminishes Warburg Effect in Lung Adenocarcinoma Cells. Front. Oncol. 2020, 10, 807. [Google Scholar] [CrossRef]
- Blasberg, R.G.; Gelovani-Tjuvajev, J. In vivo molecular-genetic imaging. J. Cell. Biochem. 2002, 39, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, F.A.; Kettunen, M.I.; Day, S.E.; Hu, D.-E.; Ardenkjaer-Larsen, J.H.; Zandt, R.I.; Jensen, P.R.; Karlsson, M.; Golman, K.; Lerche, M.H.; et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature 2008, 453, 940–943. [Google Scholar] [CrossRef]
- Jones, R.S.; Morris, M.E. Monocarboxylate transporters: Therapeutic targets and prognostic factors in disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. [Google Scholar] [CrossRef]
- Spencer, T.L.; Lehninger, A.L. L-lactate transport in Ehrlich ascites-tumour cells. Biochem. J. 1976, 154, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. Monocarboxylic acid transport. Compr. Physiol. 2013, 3, 1611–1643. [Google Scholar]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2019, 33, 48–66. [Google Scholar] [CrossRef]
- Eilertsen, M.; Andersen, S.; Al-Saad, S.; Kiselev, Y.; Donnem, T.; Stenvold, H.; Pettersen, I.; Al-Shibli, K.; Richardsen, E.; Busund, L.-T.; et al. Monocarboxylate transporters 1-4 in NSCLC: MCT1 is an independent prognostic marker for survival. PLoS ONE 2014, 9, e105038. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarug, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Milito, A.; Avnet, S.; Garcìa, A.G.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S.; Colletti, S.L.; Lovenberg, T.W.; Semple, G.; Wise, A.; Ijzerman, A.P. International union of basic and clinical pharmacology. LXXXII: Nomenclature and classification of hydroxy-carboxylic acid receptors (GPR81, GPR109A, and GPR109B). Pharmacol. Rev. 2011, 63, 269–290. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Weiszmann, J.; Reagan, J.D.; Gupte, J.; Baribault, H.; Gyuris, T.; Chen, J.-L.; Tian, H.; Li, Y. Elucidation of signaling and functional activities of an orphan GPCR, GPR81. J. Lipid Res. 2008, 49, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.; Tunaru, S.; Tang, C.; Müller, M.; Gille, A.; Sassmann, A.; Hanson, J.; Offermanns, S. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 2010, 11, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Castillo, X.; Rosafio, K.; Wyss, M.T.; Drandarov, K.; Buck, A.; Pellerin, L.; Weber, B.; Hirt, L. A probable dual mode of action for both L- and D-lactate neuroprotection in cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1561–1569. [Google Scholar] [CrossRef]
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef]
- Madaan, A.; Chaudhari, P.; Nadeau-Vallée, M.; Hamel, D.; Zhu, T.; Mitchell, G.; Samuels, M.; Pundir, S.; Dabouz, R.; Cheng, C.W.H.; et al. Müller cell-localized G-protein-coupled receptor 81 (Hydroxycarboxylic Acid Receptor 1) regulates inner retinal vasculature via Norrin/Wnt pathways. Am. J. Pathol. 2019, 189, 1878–1896. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, P.; Shanmugam, A.; Swafford, D.; Suryawanshi, A.; Bhattacharjee, P.; Hussein, M.S.; Koni, P.A.; Prasad, P.D.; Kurago, Z.B.; Thangaraju, M.; et al. GPR81, a cell-surface receptor for lactate, regulates intestinal homeostasis and protects mice from experimental colitis. J. Immunol. 2018, 200, 1781–1789. [Google Scholar] [CrossRef]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Shin, K.J.; Park, S.-A.; Park, K.S.; Park, S.; Heo, K.; Seo, Y.-K.; Noh, D.-Y.; Ryu, S.H.; Suh, P.-G. G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget 2016, 7, 70898–70911. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Park, J.Y.; Hwang, I.; Jang, B.K.; Park, K.; Kang, Y.N. Clinicopathologic significance of G protein-coupled receptor 81 in hepatocellular carcinoma. Int. J. Cancer Res. Ther. 2017, 2, 1–6. [Google Scholar]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Xie, Q.; Zhu, Z.; He, Y.; Zhang, Z.; Zhang, Y.; Wang, Y.; Luo, J.; Peng, T.; Cheng, F.; Gao, J.; et al. A lactate-induced Snail/ STAT3 pathway drives GPR81 expression in lung cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165576. [Google Scholar] [CrossRef]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [Green Version]
- Wallenius, K.; Thalén, P.; Björkman, J.-A.; Johannesson, P.; Wiseman, J.; Böttcher, G.; Fjellström, O.; Oakes, N.D. Involvement of the metabolic sensor GPR81 in cardiovascular control. JCI Insight. 2017, 2, e92564. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Li, H.; Chen, J.; Qian, Q. Lactic acid: No longer an inert and end-product of glycolysis. Physiology 2017, 32, 453–463. [Google Scholar] [CrossRef]
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Hitomi, M.; Bar-Shain, N.; Dalimov, Z.; Ellis, L.; Velpula, K.K.; Fraizer, G.C.; Gourdie, R.G.; Lathia, J.D. Connexin 43 expression is associated with increased malignancy in prostate cancer cell lines and functions to promote migration. Oncotarget 2015, 6, 11640–11651. [Google Scholar] [CrossRef]
- Dovmark, T.H.; Saccomano, M.; Hulikova, A.; Alves, F.; Swietach, P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene 2017, 36, 4538–4550. [Google Scholar] [CrossRef]
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- San-Millán, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is lactate an oncometabolite? evidence supporting a role for Lactate in the regulation of transcriptional activity of cancer-related genes in MCF7 breast cancer cells. Front. Oncol. 2020, 9, 1536. [Google Scholar] [CrossRef]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.R.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Luan, W.; Wang, Y.; Chen, X.; Shi, Y.; Wang, J.; Zhang, J.; Qian, J.; Li, R.; Tao, T.; Wei, W.; et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/ hnRNPA1 feedback loop. Oncotarget 2015, 6, 13006–13018. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [Green Version]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras (G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Mannin, B.D. The PI3K–AKT network at the interface of oncogenic signaling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenergy Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, W.; Luo, Y.; Li, H.; Xie, Y.; Wang, H.; Liu, Y.; Fan, S.; Li, Z.; Xiong, W.; et al. p53/Lactate dehydrogenase A axis negatively regulates aerobic glycolysis and tumor progression in breast cancer expressing wild-type p53. Cancer Sci. 2019, 110, 939–949. [Google Scholar] [CrossRef]
- Yang, J.; Ahmed, A.; Poon, E.; Perusinghe, N.; Brandon, A.D.H.; Box, G.; Valenti, M.; Eccles, S.; Rouschop, K.M.A.; Wouters, B.G.; et al. Small-molecule activation of p53 blocks hypoxia-inducible factor 1alpha and vascular endothelial growth factor expression in vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol. Cell. Biol. 2009, 29, 2243–2253. [Google Scholar] [CrossRef] [Green Version]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. p53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional regulation of the Warburg effect in cancer by SIX1. Cancer Cell. 2018, 33, 368–385.e7. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-X.; Yin, G.-Q.; Zhang, Z.-H.; Rong, Z.-H.; Wang, Z.-Y.; Du, D.-D.; Wang, Y.-D.; Gao, R.-X.; Xian, G.-Z. TWIST1 transcriptionally regulates glycolytic genes to promote the Warburg metabolism in pancreatic cancer. Exp. Cell Res. 2020, 386, 111713. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife 2019, 8, e50663. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.L.; Xu, Y.; Lu, X.J. Cancer immunotherapy: Challenges and clinical applications. J. Med. Genet. 2019, 56, 1–3. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, effects on immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Harmon, C.; Robinson, M.W.; Hand, F.; AlMuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Pyaram, K.; Yarosz, E.L.; Hong, H.; Lyssiotis, C.A.; Giri, S.; Chang, C.-H. Enhanced oxidative phosphorylation in NKT cells is essential for their survival and function. Proc. Natl. Acad. Sci. USA 2019, 116, 7439–7448. [Google Scholar] [CrossRef] [Green Version]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of lactate dehydrogenase-A (LDH-A) improves efficacy of anti-programmed cell death-1 (PD-1) therapy in melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, T.H. Fundamental role of dendritic cells in inducing Th2 responses. Korean J. Intern. Med. 2018, 33, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic cell reprogramming by endogenously produced lactic acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, D.; Wu, P.; Wang, Z.; Huang, J.; Gao, J.X. Serum IL-10 predicts worse outcome in cancer patients: A meta-analysis. PLoS ONE 2015, 10, e0139598. [Google Scholar] [CrossRef] [Green Version]
- Morrot, A.; Da Fonseca, L.M.; Salustiano, E.J.; Gentile, L.B.; Conde, L.; Filardy, A.A.; Franklim, T.N.; Da Costa, K.M.; Freire-De-Lima, C.G.; Freire-De-Lima, L. Metabolic symbiosis and immunomodulation: How tumor cell-derived lactate may disturb innate and adaptive immune responses. Front. Oncol. 2018, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V. Tumor cells hijack macrophages via lactic acid. Immunol. Cell Biol. 2014, 92, 647–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Brown, T.; Ramachandran, S.; Offermanns, S.; Ganapathy, V. The lactate receptor Gpr81 on non-cancer cells promotes an immunosuppressive phenotype in the tumor microenvironment. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Böhme, I.; Bosserhoff, A.K. Acidic tumor microenvironment in human melanoma pigment Cell. Melanoma Res. 2016, 29, 508–523. [Google Scholar] [CrossRef]
- Putney, L.K.; Barber, D.L. Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genom. 2004, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.; Singleton, P.A.; Diedrich, F.; Stern, R.; Gilad, E. CD44 interaction with Na + -H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J. Biol. Chem. 2004, 279, 26991–27007. [Google Scholar] [CrossRef]
- Busco, G.; Cardone, R.A.; Greco, M.R.; Bellizzi, A.; Colella, M.; Antelmi, E.; Mancini, M.T.; Dell’Aquila, M.E.; Casavola, V.; Paradiso, A.; et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010, 24, 3903–3915. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim-Hashim, A.; Abrahams, D.; Enriquez-Navas, P.M.; Luddy, K.; Gatenby, R.A.; Gillies, R.J. Tris–base buffer: A promising new inhibitor for cancer progression and metastasis. Cancer Med. 2017, 6, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Abumanhal-Masarweh, H.; Koren, L.; Zinger, A.; Yaari, Z.; Krinsky, N.; Kaneti, G.; Dahan, N.; Lupu-Haber, Y.; Suss-Toby, E.; Weiss-Messer, E.; et al. Sodium bicarbonate nanoparticles modulate the tumor pH and enhance the cellular uptake of doxorubicin. J. Control. Release. 2019, 296, 1–13. [Google Scholar] [CrossRef]
- Hamaguchi, R.; Narui, R.; Wada, H. Effects of alkalization therapy on chemotherapy outcomes in metastatic or recurrent pancreatic cancer. Anticancer Res. 2020, 40, 873–880. [Google Scholar] [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef]
- Lemma, S.; Di Pompo, G.; Porporato, P.E.; Sboarina, M.; Russell, S.; Gillies, R.J.; Baldini, N.; Sonveaux, P.; Avnet, S. MDA-MB-231 breast cancer cells fuel osteoclast metabolism and activity: A new rationale for the pathogenesis of osteolytic bone metastases. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Schwickert, G.; Walenta, S.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in human cervical cancer with incidence of metastasis. Cancer Res. 1995, 55, 4757–4759. [Google Scholar] [PubMed]
- Walenta, S.; Salameh, A.; Lyng, H.; Evensen, J.F.; Mitze, M.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am. J. Pathol. 1997, 150, 409–415. [Google Scholar]
- Walenta, S.; Chau, T.-V.; Schroeder, T.; Lehr, H.-A.; Kunz-Schughart, L.A.; Fuerst, A.; Mueller-Klieser, W. Metabolic classification of human rectal adenocarcinomas: A novel guideline for clinical oncologists? J. Cancer Res. Clin. Oncol. 2003, 129, 321–326. [Google Scholar] [CrossRef]
- Hur, H.; Xuan, Y.; Kim, Y.B.; Lee, G.; Shim, W.; Yun, J.; Ham, I.-H.; Han, S.-U. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 2013, 42, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009, 11, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hussien, R.; Oommen, S.; Gohil, K.; Brooks, G.A. Lactate sensitive transcription factor network in L6 cells: Activation of MCT1 and mitochondrial biogenesis. FASEB J. 2007, 21, 2602–2612. [Google Scholar] [CrossRef]
- Geum-Hwa, L.; Do-Sung, K.; Myung, C.J.; Soo-Wan, C.; Hyung-Ryong, K.; Han-Jung, C. Lysyl oxidase-like-1 enhances lung metastasis when lactate accumulation and monocarboxylate transporter expression are involved. Oncol. Lett. 2011, 2, 831–838. [Google Scholar]
- Pinheiro, C.; Miranda-Gonçalves, V.; Longatto-Filho, A.; Vicente, A.L.S.A.; Berardinelli, G.N.; Scapulatempo-Neto, C.; Costa, R.F.A.; Viana, C.R.; Reis, R.M.; Baltazar, M.D.F.M.; et al. The metabolic microenvironment of melanomas: Prognostic value of MCT1 and MCT4. Cell Cycle. 2016, 15, 1462–1470. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wu, M.-S.; Zou, C.; Tang, Q.; Lu, J.; Liu, D.; Wu, Y.; Yin, J.; Xie, X.; Shen, J.; et al. Downregulation of MCT1 inhibits tumor growth, metastasis and enhances chemotherapeutic efficacy in osteosarcoma through regulation of the NF- κB pathway. Cancer Lett. 2014, 342, 150–158. [Google Scholar] [CrossRef]
- Payen, V.L.; Hsu, M.Y.; Rädecke, K.S.; Wyart, E.; Vazeille, T.; Bouzin, C.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporter MCT1 promotes tumor metastasis independently of its activity as a lactate transporter. Cancer Res. 2017, 77, 5591–5601. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.C.; Nøhr-Nielsen, A.; Zeeberg, K.; Reshkin, S.J.; Hoffmann, E.K.; Novak, I.; Pedersen, S.F. Monocarboxylate transporters MCT1 and MCT4 regulate migration and invasion of pancreatic ductal adenocarcinoma cells. Pancreas 2016, 45, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.M.; Castorino, J.J.; Philp, N.J. Interaction of monocarboxylate transporter 4 with beta1-integrin and its role in cell migration. Am. J. Physiol. Cell Physiol. 2009, 296, C414–C421. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Kumar, D.; Vetrivel, U.; Parameswaran, S.; Subramanian, K.K. Structural insights on druggable hotspots in CD147: A bull’s eye view. Life Sci. 2019, 224, 76–87. [Google Scholar] [CrossRef]
- Morais-Santos, F.; Miranda-Gonçalves, V.; Pinheiro, S.; Vieira, A.F.; Paredes, J.; Schmitt, F.C.; Baltazar, F.; Pinheiro, C. Differential sensitivities to lactate transport inhibitors of breast cancer cell lines. Endocr. Relat. Cancer. 2013, 21, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.B.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. 2013, 19, 5158–5169. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Huang, X.; Xu, Z.; Dai, J.; He, H.; Zhu, Y.; Wang, H. LDHA promotes tumor metastasis by facilitating epithelial mesenchymal transition in renal cell carcinoma. Mol. Med. Rep. 2017, 16, 8335–8344. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Tomás, R. Multidrug resistance: Retrospect and prospects in anti-cancer drug treatment. Curr. Med. Chem. 2006, 13, 1859–1876. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Obre, E.; Rossignol, R. Drug discovery strategies in the field of tumor energy metabolism: Limitations by metabolic flexibility and metabolic resistance to chemotherapy. Biochim. Biophys. Acta Bioenergy 2017, 1858, 674–685. [Google Scholar] [CrossRef]
- Qu, Y.; Dou, B.; Tan, H.; Feng, Y.; Wang, N.; Wang, D. Tumor microenvironment driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer. 2019, 18, 1–16. [Google Scholar] [CrossRef]
- Kolosenko, I.; Avnet, S.; Baldini, N.; Viklund, J.; De Milito, A. Therapeutic implications of tumor interstitial acidification. Semin. Cancer Biol. 2017, 43, 119–133. [Google Scholar] [CrossRef]
- Gillies, R.; Pilot, C.; Marunaka, Y.; Fais, S. Targeting acidity in cancer and diabetes. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 273–280. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [Green Version]
- Avnet, S.; Lemma, S.; Cortini, M.; Pellegrini, P.; Perut, F.; Zini, N.; Kusuzaki, K.; Chano, T.; Grisendi, G.; Dominici, M.; et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget 2016, 7, 63408–63423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Sham, Y.Y.; Bikadi, Z.; Elmquist, W.F. pH-Dependent transport of pemetrexed by breast cancer resistance protein. Drug Metab. Dispos. 2011, 39, 1478–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.; Miéville, P.; Warren, C.M.; Saghafinia, S.; Elizabeth, A.; Peng, M.-W.; Hanahan, D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Samavati, L.; Rastogi, R.; Du, W.; Huttemann, M.; Fite, A.; Franchi, L. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol. Immunol. 2009, 46, 1867–1877. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, F.; Cui, J.Y.; Chen, L.; Chen, Y.T.; Liu, B.W. CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncol. Rep. 2018, 39, 2081–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Yao, S.; Hu, Y.; Feng, Y.; Li, M.; Bian, Z.; Zhang, J.; Qin, Y.; Qi, X.; Zhou, L.; et al. The Immune-microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin. Cancer Res. 2017, 23, 7375–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Logozzi, M.; Angelini, D.F.; Iessi, E.; Mizzoni, D.; Di Raimo, R.; Federici, C.; Lugini, L.; Borsellino, G.; Gentiluccii, A.; Pierella, F.; et al. Increased PSA expression on prostate cancer exosomes in in vitro condition and in cancer patients. Cancer Lett. 2017, 403, 318–329. [Google Scholar] [CrossRef]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; Lucas, F.A.S.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Kim, H.; Song, K.; Park, Y.; Kang, Y.; Lee, Y.; Lee, K.; Ryu, K.; Bae, J.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer. 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernández-Pérez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metab. 2018, 28, 848–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Chang, C.-Y.; Safi, R.; Liu, X.; Baldi, R.; Jasper, J.S.; Anderson, G.R.; Liu, T.; Rathmell, J.C.; Dewhirst, M.W.; et al. ERRα-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep. 2016, 15, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Quennet, V.; Yaromina, A.; Zips, D.; Rosner, A.; Walenta, S.; Baumann, M.; Mueller-Klieser, W. Tumor lactate content predicts for response to fractionated irradiation of human squamous cell carcinomas in nude mice. Radiother. Oncol. 2006, 81, 130–135. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase-A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Lea, M.A.; Guzman, Y.; Desbordes, C. Inhibition of growth by combined treatment with inhibitors of lactate dehydrogenase and either phenformin or inhibitors of 6Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase 3. Anticancer Res. 2016, 36, 1479–1488. [Google Scholar]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Baggstrom, M.Q.; Qi, Y.; Koczywas, M.; Argiris, A.; Johnson, E.A.; Millward, M.J.; Murphy, S.C.; Erlichman, C.; Rudin, C.M.; Govindan, R. A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1757–1760. [Google Scholar] [CrossRef] [Green Version]
- Heist, R.S.; Fain, J.; Chinnasami, B.; Khan, W.; Molina, J.R.; Sequist, L.V.; Temel, J.S.; Fidias, P.; Brainerd, V.; Leopold, L.; et al. Phase I/II study of AT-101 with topotecan in relapsed and refractory small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1637–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [Green Version]
- Manerba, M.; Di Ianni, L.; Govoni, M.; Roberti, M.; Recanatini, M.; Di Stefano, G. LDH inhibition impacts on heat shock response and induces senescence of hepatocellular carcinoma cells. Eur. J. Pharm. Sci. 2017, 105, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Valvona, C.J.; Fillmore, H.L. Oxamate, but not selective targeting of LDH-A, inhibits medulloblastoma cell glycolysis, growth and motility. Brain Sci. 2018, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Purkey, H.E.; Robarge, K.; Chen, J.; Chen, Z.; Corson, L.B.; Ding, C.Z.; DiPasquale, A.G.; Dragovich, P.S.; Eigenbrot, C.; Evangelista, M.; et al. Cell Active Hydroxylactam Inhibitors of Human Lactate Dehydrogenase with Oral Bioavailability in Mice. ACS Med. Chem. Lett. 2016, 7, 896–901. [Google Scholar] [CrossRef] [Green Version]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Rai, G.; Brimacombe, K.R.; Mott, B.T.; Urban, D.J.; Hu, X.; Yang, S.-M.; Lee, T.D.; Cheff, D.M.; Kouznetsova, J.; Benavides, G.A.; et al. Discovery and optimization of potent, cell active pyrazole-based inhibitors of lactate dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. [Google Scholar] [CrossRef]
- Fang, A.; Zhang, Q.; Fan, H.; Zhou, Y.; Yao, Y.; Zhang, Y.; Huang, X. Discovery of human lactate dehydrogenase A (LDHA) inhibitors as anticancer agents to inhibit the proliferation of MG-63 osteosarcoma cells. Med. Chem. Commun. 2017, 8, 1720–1726. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A novel lactate dehydrogenase inhibitor, 1-(phenylseleno)-4-(trifluoromethyl) benzene, suppresses tumor growth through apoptotic cell death. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Tao, P.; Wang, M.; Xu, P.; Lu, W.; Lei, P.; You, Q. Development of novel human lactate dehydrogenase-A inhibitors: High-throughput screening, synthesis, and biological evaluations. Eur. J. Med. Chem. 2019, 177, 105–115. [Google Scholar] [CrossRef]
- Jafary, F.; Ganjalikhany, M.R.; Moradi, A.; Hemati, M.; Jafari, S. Novel peptide inhibitors for lactate dehydrogenase A (LDHA): A survey to inhibit LDHA activity via disruption of protein-protein interaction. Sci. Rep. 2019, 9, 4686. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Guan, X.; Rodriguez-Cruz, V.; Morris, M.E. Cellular Uptake of MCT1 Inhibitors AR-C155858 and AZD3965 and Their Effects on MCT-Mediated Transport of L-Lactate in Murine 4T1 Breast Tumor Cancer Cells. AAPS J. 2019, 21, 13. [Google Scholar] [CrossRef]
- Doherty, J.R.; Yang, C.; Scott, K.E.N.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Polański, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.-M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058. [Google Scholar] [CrossRef] [Green Version]
- Ždralević, M.; Vučetić, M.; Daher, B.; Marchiq, I.; Parks, S.K.; Pouysségur, J. Disrupting the ‘Warburg effect’ re-routes cancer cells to OXPHOS offering a vulnerability point via ‘ferroptosis’-induced cell death. Adv. Biol. Regul. 2018, 68, 55–63. [Google Scholar] [CrossRef]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Sanità, P.; Capulli, M.; Teti, A.; Galatioto, G.P.; Vicentini, C.; Chiarugi, P.; Bologna, M.; Angelucci, A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer. 2014, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201e207. [Google Scholar] [CrossRef]
- Quanz, M.; Bender, E.; Kopitz, C.; Grünewald, S.; Schlicker, A.; Schwede, W.; Eheim, A.; Toschi, L.; Neuhaus, R.; Richter, C.; et al. Preclinical efficacy of the novel monocarboxylate transporter 1 inhibitor BAY-8002 and associated markers of resistance. Mol. Cancer Ther. 2018, 17, 2285–2296. [Google Scholar] [CrossRef] [Green Version]
- Marchiq, I.; Le Floch, R.; Roux, D.; Simon, M.P.; Pouyssegur, J. Genetic disruption of lactate/Hþ symporters (MCTs) and their subunit CD147/ BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [Green Version]
- Follman, K.E.; Morris, M.E. Treatment of γ-hydroxybutyric acid (GHB) and γ-butyrolactone (GBL) overdose with two potent monocarboxylate transporter 1 (MCT1) inhibitors, AZD3965 and AR-C155858. J. Pharmacol. Exp. Ther. 2019, 370, 84–91. [Google Scholar] [CrossRef]
- Hao, J.; Chen, H.; Madigan, M.C.; Cozzi, P.J.; Beretov, J.; Xiao, W.; Delprado, W.J.; Russell, P.J.; Li, Y. Co-expression of CD147 (EMMPRIN), CD44v3-10, MDR1 and monocarboxylate transporters is associated with prostate cancer drug resistance and progression. Br. J. Cancer. 2010, 103, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Le Fur, Y.; Vilmen, C.; Frassineti, F.; Daniel, L.; Halestrap, A.P.; Cozzone, P.J.; Pouysségur, J.; Lutz, N.W. In vivo pH in metabolic-defective Ras-transformed fibroblast tumors: Key role of the monocarboxylate transporter, MCT4, for inducing an alkaline intracellular pH. Int. J Cancer. 2012, 130, 1511–1520. [Google Scholar] [CrossRef]
- Sasaki, S.; Futagi, Y.; Ideno, M.; Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Effect of diclofenac on SLC16A3/MCT4 by the Caco-2 cell line. Drug Metab. Pharmacokinet. 2016, 31, 218–223. [Google Scholar] [CrossRef]
- Futagi, Y.; Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Identification of a selective inhibitor of human monocarboxylate transporter 4. Biochem. Biophys. Res. Commun. 2018, 495, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Marchiq, I.; Pouyssegur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Nancolas, B.; Guo, L.; Zhou, R.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Blair, I.A.; Glickson, J.D.; Halestrap, A.P. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem. J. 2016, 473, 929–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wen, Z.; Xie, J.; Zhao, Y.; Zhao, L.; Zhang, S.; Liu, Y.; Xue, Y.; Shi, M. MACC1 mediates chemotherapy sensitivity of 5-FU and cisplatin via regulating MCT1 expression in gastric cancer. Biochem. Biophys. Res. Commun. 2017, 485, 665–671. [Google Scholar] [CrossRef]
- Wilson, M.C.; Meredith, D.; Fox, J.E.M.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.-G.; Wang, L.; Cui, H.-Y.; Peng, J.-L.; Wang, S.-J.; Geng, J.-J.; Liu, J.-D.; Feng, F.; Song, F.; Li, L.; et al. A novel small-molecule compound targeting CD147 inhibits the motility and invasion of hepatocellular carcinoma cells. Oncotarget 2016, 7, 9429–9447. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Y.; Sun, Q.; Feng, F.; Hube, M.; Mi, L.; Chen, Z. Preclinical pharmacokinetics, tolerability, and pharmacodynamics of metuzumab, a novel CD147 human mouse chimeric and glycol engineered antibody. Mol. Cancer Ther. 2015, 14, 162–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Type of Cancer or Cell/Animal Model | Research Phase | References |
|---|---|---|---|---|
| LDH | FX-11 | B-lymphoid cells (P493, P198) Xenograft model | Pre-clinical | [167] |
| Gossypol AT-10 | Multiple kinds of cancer | Phase I and Phase II clinical trialsa | [171,172] | |
| Galloflavin | Liver cancer (PLC/PRF/5) Hepatocellular carcinoma | Pre-clinical | [168,174] | |
| N-hydroxyindole-based compounds | Colon (Caco-2, HCT116 and HT29) Bladder (5637, HT1197, HT1376, RT4, SW780, T24, TCCSUP and UM-UC-3) | Pre-clinical | [169] | |
| FX866 | Pancreatic cancer (P198) Xenograft model | Pre-clinical | [167] | |
| Oxamate | Hepatocellular carcinoma Medulloblastoma | Pre-clinical | [174,175] | |
| 2 Thio-6-oxo1,6-dihydropyrinidine (DHPMs) | Pancreatic carcinoma (MIA PaCa-2) Mouse model | Pre-clinical | [176] | |
| GNE-140 | Colon adenocarcinoma (LS174T) Mouse model Pancreatic carcinoma (MIA PaCa-2) | Pre-clinical | [177,178] | |
| Pyzazole based inhibitors | Pancreatic carcinoma (MIA PaCa-2) A673 Sarcoma (A673) | Pre-clinical | [179] | |
| 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene (PSTMP) | Large cell lung cancer (NCI-H460) Breast cancer (MCF-7) Hepatocellular carcinoma (Hep3B) Malignan melanoma (A375) Colorectal adenocarcinoma (HT29) Murine lung cancer (LLC) | Pre-clinical | [180] | |
| Compound 11 | Osteosarcoma (MG-63) | Pre-clinical | [181] | |
| Compound 24c | Pancreas carcinoma (MiaPaCa-2) | Pre-clinical | [182] | |
| Peptides collections (QLYNL, LIYNLL, IYNLLK, KWYNVA, and KVVYNV) | None | In silico modeling | [183] |
| Target | Drug | Type of Cancer or Cell/Animal Model | Research Phase | References |
|---|---|---|---|---|
| MCT1 | AR-C155858 | Murine breast cancer (4T1) | Pre-clinical | [186] |
| SR 13800 | Burkitt lymphoma (Raji) | Pre-clinical | [185] | |
| AZD 3965 | Human diffuse large B-cell lymphomas (HBL-1 and TMD8) Human B-cell lymphoma (WSU-CLCL-2 and SU DHL10) Lymphoblast (HT) B-cell non-Hodgkin lymphoma (Karpas-422 NHL) Raji Burkitt’s lymphoma cells | Pre-clinical Phase I/II of clinical trials a. | [186,188,189] | |
| α- cyano-4-hydroxycinnamate (CHC) | Colorectal cancer (HCT15 and RKO) Murine cancer model | Pre-clinical | [82,192] | |
| BAY-8002 | Hematopoietic malignancies, Raji, and Daudi Burkitt lymphoma cells | Pre-clinical | [193] | |
| MTC4 | Diclofenac | Caco-2 cell line | FDA- Approved as anti-inflammatory drug | [200] |
| Bindarit | Xenopus oocyte | Experimental Research | [201] | |
| AZ93 | Wide range of cancer cells | Pre-clinical | [202] | |
| MCT1/MCT4 | Syrosingopine | HeLa, HAP1, HL60 cells, liver tumor mouse model | Pre-clinical | [189] |
| Lonidamine | DB-1 melanoma cell | Pre-clinical Phase III of clinical trials (prostate cancer) b | [203] | |
| CD147 | pCMBS | Molecular biology (Xenopus oocyte, murine cells) | Experimental Research | [205] |
| AC-73 | Hepatocellular carcinoma (SMMC-7721, Huh7) Orthotopic transplant nude mouse model | Pre-clinical | [206] | |
| Metuzumab | Xenograft models (A549, NCI-H520) Monkey model | Pre-clinical | [207] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Tomás, R.; Pérez-Guillén, I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244. https://doi.org/10.3390/cancers12113244
Pérez-Tomás R, Pérez-Guillén I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers. 2020; 12(11):3244. https://doi.org/10.3390/cancers12113244
Chicago/Turabian StylePérez-Tomás, Ricardo, and Isabel Pérez-Guillén. 2020. "Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment" Cancers 12, no. 11: 3244. https://doi.org/10.3390/cancers12113244
APA StylePérez-Tomás, R., & Pérez-Guillén, I. (2020). Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers, 12(11), 3244. https://doi.org/10.3390/cancers12113244