HOX Genes Family and Cancer: A Novel Role for Homeobox B9 in the Resistance to Anti-Angiogenic Therapies


Abstract
:Simple Summary
Abstract
1. Introduction
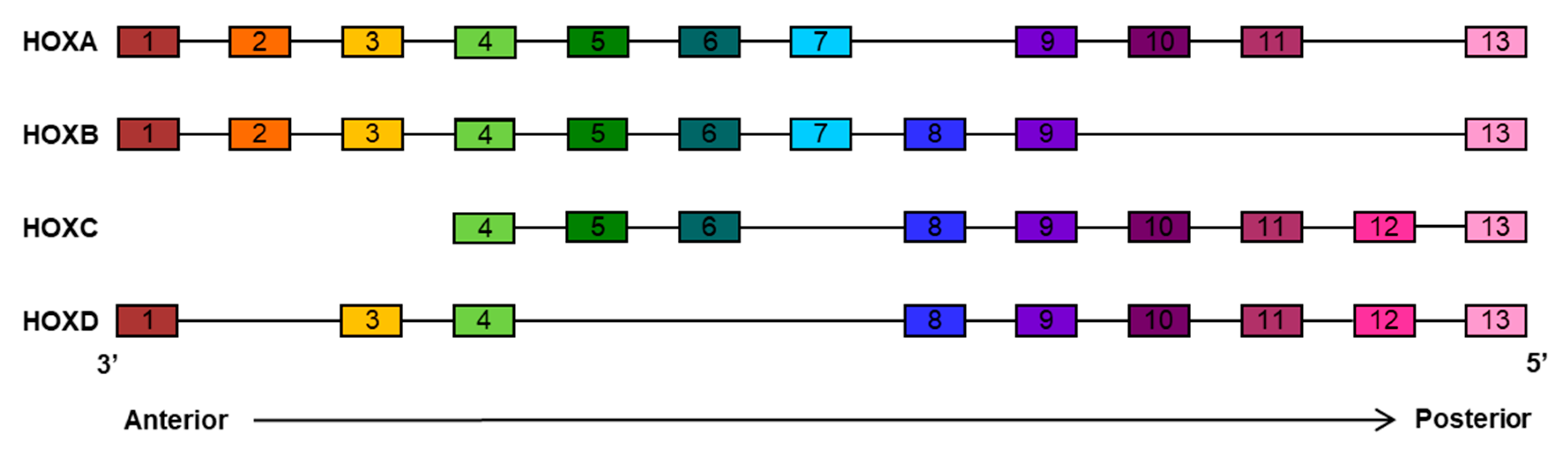
2. HOX Family Transcription Factors
2.1. HOX Family in Angiogenesis
3. HOXB9 in Cancer
3.1. HOXB9 Regulation in Cancer
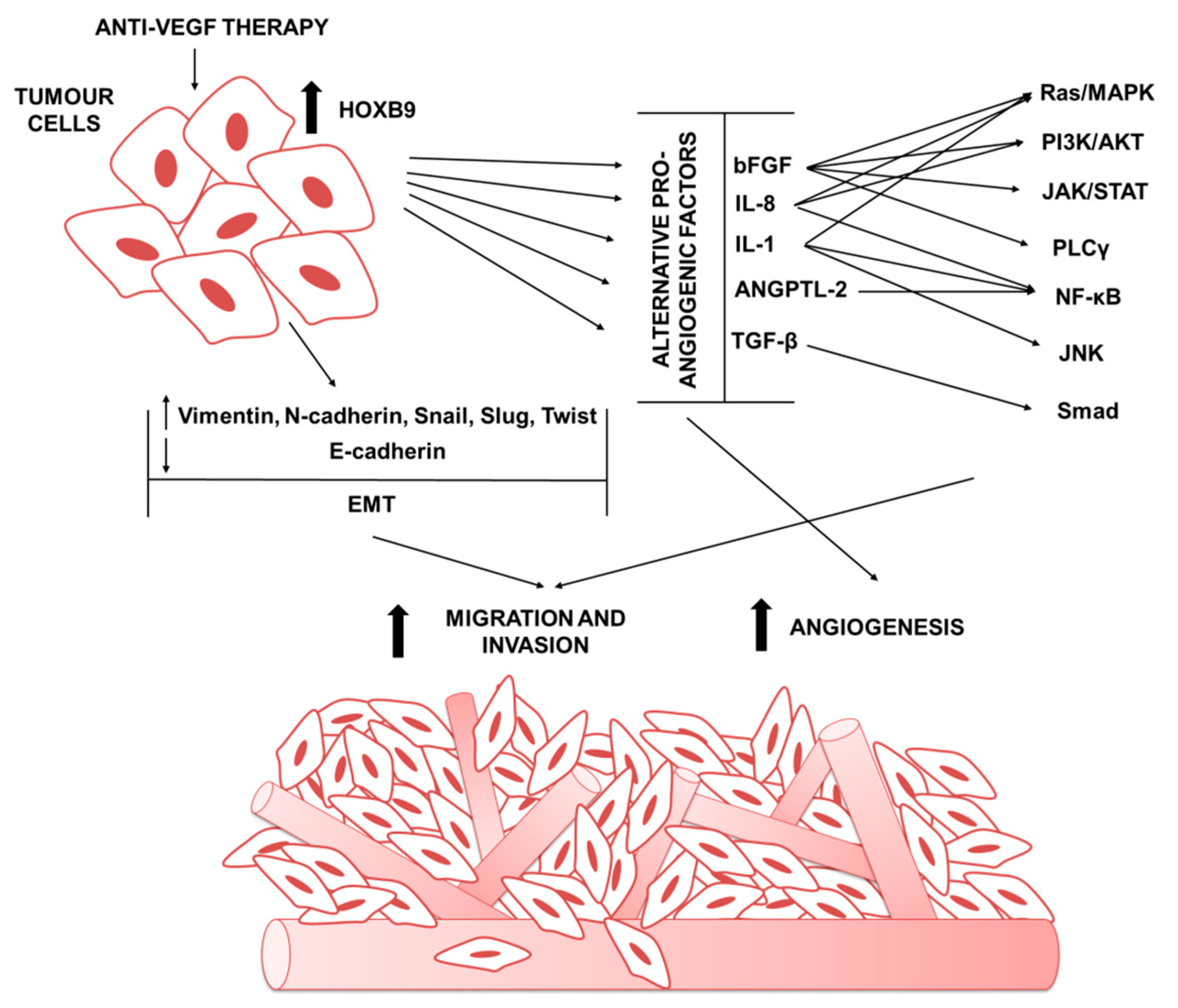
4. The Role of HOXB9 in Tumor Anti-Angiogenic Treatments Escape
4.1. HOXB9 in Mediating the Expression of Alternative Pro-Angiogenic Factors
4.2. HOXB9 Role in Tumor Invasivenes and Metastasis
4.3. HOXB9 in Modulating Stromal Cell in Tumor Microenvirment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tortora, G.; Melisi, D.; Ciardiello, F. Angiogenesis: A target for cancer therapy. Curr. Pharm. Des. 2004, 10, 11–26. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Hillan, K.J.; Gerber, H.-P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Garon, E.B.; Ciuleanu, T.-E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Berlin, J.; Chan, E. Aflibercept. Clin. Cancer Res. 2013, 19, 1920–1925. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.; Kerbel, R.S. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.d.M.; Wijendran, V.; Shioda, T. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.; Wang, P.; Niu, M.; Wang, Y.; Zhu, X.; Guo, Y.; Zhang, H. High expression of transcriptional factor HoxB9 predicts poor prognosis in patients with lung adenocarcinoma. Histopathology 2015, 66, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Chiba, N.; Ozawa, Y.; Hikita, K.; Okihara, M.; Sano, T.; Tomita, K.; Takano, K.; Kawachi, S. Increased expression of HOXB9 in hepatocellular carcinoma predicts poor overall survival but a beneficial response to sorafenib. Oncol. Rep. 2017, 37, 2270–2276. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Simionato, F.; Ligorio, F.; Cremolini, C.; Loupakis, F.; Alì, G.; Rossini, D.; Merz, V.; Santoro, R. Homeobox B9 mediates resistance to anti-VEGF therapy in colorectal cancer patients. Clin. Cancer Res. 2017, 23, 4312–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, C.B. Current maps of the location of the mutant genes of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1921, 7, 127. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. In Genes, Development and Cancer; Springer: Berlin/Heidelberg, Germany, 1978; pp. 205–217. [Google Scholar]
- Scott, M.P. Vertebrate homeobox gene nomenclature. Cell 1992, 71, 551–553. [Google Scholar] [CrossRef]
- Levine, M.; Hoey, T. Homeobox proteins as sequence-specific transcription factors. Cell 1988, 55, 537–540. [Google Scholar] [CrossRef]
- McGinnis, W.; Krumlauf, R. Homeobox genes and axial patterning. Cell 1992, 68, 283–302. [Google Scholar] [CrossRef]
- Mann, R.S.; Lelli, K.M.; Joshi, R. Hox specificity: Unique roles for cofactors and collaborators. Curr. Top. Dev. Biol. 2009, 88, 63–101. [Google Scholar]
- Iimura, T.; Pourquié, O. Hox genes in time and space during vertebrate body formation. Dev. Growth Differ. 2007, 49, 265–275. [Google Scholar] [CrossRef]
- Horan, G.S.; Kovàcs, E.N.; Behringer, R.R.; Featherstone, M.S. Mutations in paralogous Hox genes result in overlapping homeotic transformations of the axial skeleton: Evidence for unique and redundant function. Dev. Biol. 1995, 169, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, J.M.; Puetz, J.; Thomas, K.R.; Capecchi, M.R. Maintenance of functional equivalence during paralogous Hox gene evolution. Nature 2000, 403, 661–665. [Google Scholar] [CrossRef]
- Mansour, M.A.; Senga, T. HOXD8 exerts a tumor-suppressing role in colorectal cancer as an apoptotic inducer. Int. J. Biochem. Cell Biol. 2017, 88, 1–13. [Google Scholar] [CrossRef] [Green Version]
- De Kumar, B.; Parker, H.J.; Paulson, A.; Parrish, M.E.; Zeitlinger, J.; Krumlauf, R. Hoxa1 targets signaling pathways during neural differentiation of ES cells and mouse embryogenesis. Dev. Biol. 2017, 432, 151–164. [Google Scholar] [CrossRef]
- Tsuboi, M.; Taniuchi, K.; Shimizu, T.; Saito, M.; Saibara, T. The transcription factor HOXB7 regulates ERK kinase activity and thereby stimulates the motility and invasiveness of pancreatic cancer cells. J. Biol. Chem. 2017, 292, 17681–17702. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Rupaimoole, R.; Shen, F.; Pradeep, S.; Pecot, C.V.; Ivan, C.; Nagaraja, A.S.; Gharpure, K.M.; Pham, E.; Hatakeyama, H. A miR-192-EGR1-HOXB9 regulatory network controls the angiogenic switch in cancer. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Boudreau, N.; Andrews, C.; Srebrow, A.; Ravanpay, A.; Cheresh, D.A. Induction of the angiogenic phenotype by Hox D3. J. Cell Biol. 1997, 139, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Chisaka, O.; Kameda, Y. Hoxa3 regulates the proliferation and differentiation of the third pharyngeal arch mesenchyme in mice. Cell Tissue Res. 2005, 320, 77–89. [Google Scholar] [CrossRef]
- Myers, C.; Charboneau, A.; Boudreau, N. Homeobox B3 promotes capillary morphogenesis and angiogenesis. J. Cell Biol. 2000, 148, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Bruhl, T.; Urbich, C.; Aicher, D.; Acker-Palmer, A.; Zeiher, A.M.; Dimmeler, S. Homeobox A9 transcriptionally regulates the EphB4 receptor to modulate endothelial cell migration and tube formation. Circ. Res. 2004, 94, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Rössig, L.; Urbich, C.; Brühl, T.; Dernbach, E.; Heeschen, C.; Chavakis, E.; Sasaki, K.-I.; Aicher, D.; Diehl, F.; Seeger, F. Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J. Exp. Med. 2005, 201, 1825–1835. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Moser, M.; Bautch, V.L.; Patterson, C. HoxB5 is an upstream transcriptional switch for differentiation of the vascular endothelium from precursor cells. Mol. Cell. Biol. 2003, 23, 5680–5691. [Google Scholar] [CrossRef] [Green Version]
- Carè, A.; Silvani, A.; Meccia, E.; Mattia, G.; Stoppacciaro, A.; Parmiani, G.; Peschle, C.; Colombo, M.P. HOXB7 constitutively activates basic fibroblast growth factor in melanomas. Mol. Cell. Biol. 1996, 16, 4842–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carè, A.; Felicetti, F.; Meccia, E.; Bottero, L.; Parenza, M.; Stoppacciaro, A.; Peschle, C.; Colombo, M.P. HOXB7: A key factor for tumor-associated angiogenic switch. Cancer Res. 2001, 61, 6532–6539. [Google Scholar]
- Tan, Z.; Chen, K.; Wu, W.; Zhou, Y.; Zhu, J.; Wu, G.; Cao, L.; Zhang, X.; Guan, H.; Yang, Y. Overexpression of HOXC10 promotes angiogenesis in human glioma via interaction with PRMT5 and upregulation of VEGFA expression. Theranostics 2018, 8, 5143. [Google Scholar] [CrossRef]
- Rhoads, K.; Arderiu, G.; Charboneau, A.; Hansen, S.L.; Hoffman, W.; Boudreau, N. A role for Hox A5 in regulating angiogenesis and vascular patterning. Lymphat. Res. Biol. 2005, 3, 240–252. [Google Scholar] [CrossRef]
- Arderiu, G.; Cuevas, I.; Chen, A.; Carrio, M.; East, L.; Boudreau, N.J. HoxA5 stabilizes adherens junctions via increased Akt1. Cell Adhes. Migr. 2007, 1, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Myers, C.; Charboneau, A.; Cheung, I.; Hanks, D.; Boudreau, N. Sustained expression of homeobox D10 inhibits angiogenesis. Am. J. Pathol. 2002, 161, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Carrio, M.; Arderiu, G.; Myers, C.; Boudreau, N.J. Homeobox D10 induces phenotypic reversion of breast tumor cells in a three-dimensional culture model. Cancer Res. 2005, 65, 7177–7185. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Cuevas, I.; Kenny, P.A.; Miyake, H.; Mace, K.; Ghajar, C.; Boudreau, A.; Bissell, M.; Boudreau, N. Endothelial cell migration and vascular endothelial growth factor expression are the result of loss of breast tissue polarity. Cancer Res. 2009, 69, 6721–6729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromental-Ramain, C.; Warot, X.; Lakkaraju, S.; Favier, B.; Haack, H.; Birling, C.; Dierich, A.; Chambon, P. Specific and redundant functions of the paralogous Hoxa-9 and Hoxd-9 genes in forelimb and axial skeleton patterning. Development 1996, 122, 461–472. [Google Scholar]
- Xu, B.; Wellik, D.M. Axial Hox9 activity establishes the posterior field in the developing forelimb. Proc. Natl. Acad. Sci. USA 2011, 108, 4888–4891. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Capecchi, M.R. Paralogous mouse Hox genes, Hoxa9, Hoxb9, and Hoxd9, function together to control development of the mammary gland in response to pregnancy. Proc. Natl. Acad. Sci. USA 1999, 96, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Magli, M.C.; Largman, C.; Lawrence, H.J. Effects of HOX homeobox genes in blood cell differentiation. J. Cell. Physiol. 1997, 173, 168–177. [Google Scholar] [CrossRef]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. HOX genes and their role in the development of human cancers. J. Mol. Med. 2014, 92, 811–823. [Google Scholar] [CrossRef]
- Huang, K.; Yuan, R.; Wang, K.; Hu, J.; Huang, Z.; Yan, C.; Shen, W.; Shao, J. Overexpression of HOXB9 promotes metastasis and indicates poor prognosis in colon cancer. Chin. J. Cancer Res. 2014, 26, 72. [Google Scholar]
- Seki, H.; Hayashida, T.; Jinno, H.; Hirose, S.; Sakata, M.; Takahashi, M.; Maheswaran, S.; Mukai, M.; Kitagawa, Y. HOXB9 expression promoting tumor cell proliferation and angiogenesis is associated with clinical outcomes in breast cancer patients. Ann. Surg. Oncol. 2012, 19, 1831–1840. [Google Scholar] [CrossRef]
- Fang, L.; Xu, Y.; Zou, L. Overexpressed homeobox B9 regulates oncogenic activities by transforming growh factor-β1 in gliomas. Biochem. Biophys. Res. Commun. 2014, 446, 272–279. [Google Scholar] [CrossRef]
- Kato, F.; Wada, N.; Hayashida, T.; Fukuda, K.; Nakamura, R.; Takahashi, T.; Kawakubo, H.; Takeuchi, H.; Kitagawa, Y. Experimental and clinicopathological analysis of HOXB9 in gastric cancer. Oncol. Lett. 2019, 17, 3097–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, S.; Gu, Y.; Xu, B.; Hu, H.; Yang, Y.; Kong, X.; Wu, K. Decreased expression of HOXB9 is related to poor overall survival in patients with gastric carcinoma. Dig. Liver Dis. 2013, 45, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Ansari, K.I.; Bhan, A.; Kasiri, S.; Hussain, I.; Mandal, S.S. Homeodomain-containing protein HOXB 9 regulates expression of growth and angiogenic factors, facilitates tumor growth in vitro and is overexpressed in breast cancer tissue. FEBS J. 2012, 279, 3715–3726. [Google Scholar] [CrossRef]
- Chiba, N.; Comaills, V.; Shiotani, B.; Takahashi, F.; Shimada, T.; Tajima, K.; Winokur, D.; Hayashida, T.; Willers, H.; Brachtel, E. Homeobox B9 induces epithelial-to-mesenchymal transition-associated radioresistance by accelerating DNA damage responses. Proc. Natl. Acad. Sci. USA 2012, 109, 2760–2765. [Google Scholar] [CrossRef] [Green Version]
- Zhussupova, A.; Hayashida, T.; Takahashi, M.; Miyao, K.; Okazaki, H.; Jinno, H.; Kitagawa, Y. An E2F1-HOXB9 transcriptional circuit is associated with breast cancer progression. PLoS ONE 2014, 9, e105285. [Google Scholar] [CrossRef]
- Hoshino, Y.; Hayashida, T.; Hirata, A.; Takahashi, H.; Chiba, N.; Ohmura, M.; Wakui, M.; Jinno, H.; Hasegawa, H.; Maheswaran, S. Bevacizumab terminates homeobox B9-induced tumor proliferation by silencing microenvironmental communication. Mol. Cancer 2014, 13, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Xu, W.; Zhan, J.; Ma, J.; Li, X.; Xie, Y.; Wang, J.; Zhu, W.-G.; Luo, J.; Zhang, H. PCAF-mediated acetylation of transcriptional factor HOXB9 suppresses lung adenocarcinoma progression by targeting oncogenic protein JMJD6. Nucleic Acids Res. 2016, 44, 10662–10675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Liu, H.; Feng, Q.; Liu, J.; Ming, L. HOXB9 promotes endometrial cancer progression by targeting E2F3. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, Q.; He, C.; Liang, D.; Yi, Q.; Shi, J.; Wan, B.; Yang, R.; Li, L.; Sha, S. HOXB9 inhibits proliferation in gastric carcinoma cells via suppression of phosphorylated-Akt and NF-κB-dependent Snail expression. Dig. Liver Dis. 2019, 51, 157–165. [Google Scholar] [CrossRef]
- Sha, L.; Dong, L.; Lv, L.; Bai, L.; Ji, X. HOXB9 promotes epithelial-to-mesenchymal transition via transforming growth factor-β1 pathway in hepatocellular carcinoma cells. Clin. Exp. Med. 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Li, F.; Dong, L.; Xing, R.; Wang, L.; Luan, F.; Yao, C.; Ji, X.; Bai, L. Homeobox B9 is overexpressed in hepatocellular carcinomas and promotes tumor cell proliferation both in vitro and in vivo. Biochem. Biophys. Res. Commun. 2014, 444, 241–247. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.-F.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massagué, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009, 138, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.-S.; Oh, E.; Park, J.-R.; Lee, J.-S.; Bae, G.-Y.; Koo, J.-H.; Kim, H.; Choi, Y.-L.; Choi, Y.S.; Kim, J. GalNAc-T14 promotes metastasis through Wnt dependent HOXB9 expression in lung adenocarcinoma. Oncotarget 2015, 6, 41916. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Zhu, F.-Y.; Chen, L.; Wang, K. HoxB9 promotes the migration and invasion via TGF-β1/Smad2/Slug signaling pathway in oral squamous cell carcinoma. Am. J. Transl. Res. 2017, 9, 1151. [Google Scholar]
- Sun, C.; Han, C.; Wang, P.; Jin, Y.; Sun, Y.; Qu, L. HOXB9 expression correlates with histological grade and prognosis in LSCC. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Sun, X.; Song, J.; Zhang, J.; Zhan, J.; Fang, W.; Zhang, H. Acetylated HOXB9 at lysine 27 is of differential diagnostic value in patients with pancreatic ductal adenocarcinoma. Front. Med. 2020, 14, 91–100. [Google Scholar] [CrossRef]
- Xu, H.; Wu, S.; Shen, X.; Wu, D.; Qin, Z.; Wang, H.; Chen, X.; Sun, X. Silencing of HOXB9 suppresses cellular proliferation, angiogenesis, migration and invasion of prostate cancer cells. J. Biosci. 2020, 45, 40. [Google Scholar] [CrossRef]
- Hatzis, P.; van der Flier, L.G.; van Driel, M.A.; Guryev, V.; Nielsen, F.; Denissov, S.; Nijman, I.J.; Koster, J.; Santo, E.E.; Welboren, W. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 2008, 28, 2732–2744. [Google Scholar] [CrossRef] [Green Version]
- Taylor, H.S. The role of HOX genes in human implantation. Hum. Reprod. Update 2000, 6, 75–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, K.I.; Shrestha, B.; Hussain, I.; Kasiri, S.; Mandal, S.S. Histone methylases MLL1 and MLL3 coordinate with estrogen receptors in estrogen-mediated HOXB9 expression. Biochemistry 2011, 50, 3517–3527. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, T.; Xu, W.; Wang, P.; Wan, J.; Wang, Y.; Zhan, J.; Zhang, H. HOXB9 acetylation at K27 is responsible for its suppression of colon cancer progression. Cancer Lett. 2018, 426, 63–72. [Google Scholar] [CrossRef]
- Miller, K.D.; Sweeney, C.J.; Sledge, G.W. Can tumor angiogenesis be inhibited without resistance. In Mechanisms of Angiogenesis; Springer: Berlin/Heidelberg, Germany, 2005; pp. 95–112. [Google Scholar]
- Ruegg, C.; Mutter, N. Anti-angiogenic therapies in cancer: Achievements and open questions. Bull. Cancer 2007, 94, 753–762. [Google Scholar]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Hayashida, T.; Jinno, H.; Seki, H.; Takahashi, M.; Sakata, M.; Hirose, S.; Mukai, M.; Kitagawa, Y. The relationship of HOXB9 expression promoting tumor cell proliferation and angiogenesis to clinical outcomes of patients with breast cancer. J. Clin. Oncol. 2011, 29, 10546. [Google Scholar] [CrossRef]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Eswarakumar, V.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. 2010, 28, 453. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.-T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.-J.; Zhu, M. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, C.; Moccia, T.; Zhu, C.; Paradiso, G.; Budillon, A.; Chiao, P.J.; Abbruzzese, J.L.; Melisi, D. Anti-VEGF treatment-resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an EMT cell phenotype. Clin. Cancer Res. 2011, 17, 5822–5832. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, C.; Sanmamed, M.F.; Rodríguez-Ruiz, M.E.; Teijeira, Á.; Oñate, C.; González, Á.; Ponz, M.; Schalper, K.A.; Pérez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef]
- Merz, V.; Zecchetto, C.; Santoro, R.; Simionato, F.; Sabbadini, F.; Mangiameli, D.; Piro, G.; Cavaliere, A.; Deiana, M.; Valenti, M.T.; et al. Plasma IL8 Is a Biomarker for TAK1 Activation and Predicts Resistance to Nanoliposomal Irinotecan in Patients with Gemcitabine-Refractory Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4661–4669. [Google Scholar] [CrossRef]
- Gyanchandani, R.; Sano, D.; Alves, M.V.O.; Klein, J.D.; Knapick, B.A.; Oh, S.; Myers, J.N.; Kim, S. Interleukin-8 as a modulator of response to bevacizumab in preclinical models of head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 761–770. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.-N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchelor, T.T.; Gerstner, E.R.; Emblem, K.E.; Duda, D.G.; Kalpathy-Cramer, J.; Snuderl, M.; Ancukiewicz, M.; Polaskova, P.; Pinho, M.C.; Jennings, D. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc. Natl. Acad. Sci. USA 2013, 110, 19059–19064. [Google Scholar] [CrossRef] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Nussinov, R.; Keskin, O. The structural pathway of interleukin 1 (IL-1) initiated signaling reveals mechanisms of oncogenic mutations and SNPs in inflammation and cancer. PLoS Comput. Biol. 2014, 10, e1003470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, C.; Melisi, D. NF-kappaB as a target for pancreatic cancer therapy. Expert Opin. Targets 2012, 16 (Suppl. 2), S1–S10. [Google Scholar]
- Melisi, D.; Chiao, P.J. NF-kappa B as a target for cancer therapy. Expert Opin. Targets 2007, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Niu, J.; Chang, Z.; Xia, Q.; Peng, B.; Ishiyama, S.; Evans, D.B.; Chiao, P.J. Secreted interleukin-1alpha induces a metastatic phenotype in pancreatic cancer by sustaining a constitutive activation of nuclear factor-kappaB. Mol. Cancer Res. 2009, 7, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Ju, H.Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-kappaB Activation. Clin. Cancer Res. 2016, 22, 1432–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K. Proinflammatory cytokine IL-1β promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: In vivo analysis of tumor-stromal interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, C.; Tamburrino, A.; Piro, G.; Boschi, F.; Cataldo, I.; Zanotto, M.; Mina, M.M.; Zanini, S.; Sbarbati, A.; Scarpa, A.; et al. Combined inhibition of IL1, CXCR1/2, and TGFbeta signaling pathways modulates in-vivo resistance to anti-VEGF treatment. Anticancer Drugs 2016, 27, 29–40. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Liu, Z.; Ten Dijke, P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFbeta receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Melisi, D.; Hollebecque, A.; Oh do, Y.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Simionato, F.; Park, O.J.; Bendell, J.; et al. A Phase 1b Dose-Escalation and Cohort-Expansion Study of Safety and Activity of the Transforming Growth Factor (TGF) β Receptor I Kinase Inhibitor Galunisertib Plus the Anti-PD-L1 Antibody Durvalumab in Metastatic Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 4124. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef] [Green Version]
- Piro, G.; Giacopuzzi, S.; Bencivenga, M.; Carbone, C.; Verlato, G.; Frizziero, M.; Zanotto, M.; Mina, M.M.; Merz, V.; Santoro, R.; et al. TAK1-regulated expression of BIRC3 predicts resistance to preoperative chemoradiotherapy in oesophageal adenocarcinoma patients. Br. J. Cancer 2015, 113, 878–885. [Google Scholar] [CrossRef]
- Santoro, R.; Carbone, C.; Piro, G.; Chiao, P.J.; Melisi, D. TAK-ing aim at chemoresistance: The emerging role of MAP3K7 as a target for cancer therapy. Drug Resist. Updates 2017, 33–35, 36–42. [Google Scholar] [CrossRef]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 expression inhibits YAP and TAZ oncogenic functions in pancreatic cancer. Mol. Cancer 2019, 19, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.I.; Pietras, K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 2011, 117, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Bertolino, P.; Deckers, M.; Lebrin, F.; ten Dijke, P. Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest 2005, 128, 585S–590S. [Google Scholar] [CrossRef]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-L.; Chen, Z.-Q.; Zhu, S.-L.; Liu, T.-W.; Wen, Y.; Su, Y.-S.; Xi, X.-J.; Hu, Y.; Lian, L.; Liu, F.-B. Prognostic value of transforming growth factor-beta in patients with colorectal cancer who undergo surgery: A meta-analysis. BMC Cancer 2017, 17, 240. [Google Scholar] [CrossRef]
- Dave, H.; Shah, M.; Trivedi, S.; Shukla, S. Prognostic utility of circulating transforming growth factor beta 1 in breast cancer patients. Int. J. Biol. Markers 2012, 27, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Piao, Y.; Jeong, K.J.; Dong, J.; de Groot, J.F. Periostin (POSTN) regulates tumor resistance to antiangiogenic therapy in glioma models. Mol. Cancer Ther. 2016, 15, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Bockhorn, M.; Tsuzuki, Y.; Xu, L.; Frilling, A.; Broelsch, C.E.; Fukumura, D. Differential vascular and transcriptional responses to anti-vascular endothelial growth factor antibody in orthotopic human pancreatic cancer xenografts. Clin. Cancer Res. 2003, 9, 4221–4226. [Google Scholar] [PubMed]
- Carbone, C.; Piro, G.; Merz, V.; Simionato, F.; Santoro, R.; Zecchetto, C.; Tortora, G.; Melisi, D. Angiopoietin-Like Proteins in Angiogenesis, Inflammation and Cancer. Int. J. Mol. Sci. 2018, 19, 431. [Google Scholar] [CrossRef] [Green Version]
- Aoi, J.; Endo, M.; Kadomatsu, T.; Miyata, K.; Nakano, M.; Horiguchi, H.; Ogata, A.; Odagiri, H.; Yano, M.; Araki, K. Angiopoietin-like protein 2 is an important facilitator of inflammatory carcinogenesis and metastasis. Cancer Res. 2011, 71, 7502–7512. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Nakano, M.; Kadomatsu, T.; Fukuhara, S.; Kuroda, H.; Mikami, S.; Hato, T.; Aoi, J.; Horiguchi, H.; Miyata, K. Tumor Cell-Derived Angiopoietin-like Protein ANGPTL2 Is a Critical Driver of Metastasis. Cancer Res. 2012, 72, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Carbone, C.; Piro, G.; Fassan, M.; Tamburrino, A.; Mina, M.M.; Zanotto, M.; Chiao, P.J.; Bassi, C.; Scarpa, A.; Tortora, G.; et al. An angiopoietin-like protein 2 autocrine signaling promotes EMT during pancreatic ductal carcinogenesis. Oncotarget 2015, 6, 13822–13834. [Google Scholar] [CrossRef]
- Hato, T.; Tabata, M.; Oike, Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med. 2008, 18, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Oike, Y.; Satoh, S.; Tabata, Y.; Niikura, Y.; Morisada, T.; Akao, M.; Urano, T.; Ito, Y.; Miyamoto, T. Cooperative interaction of Angiopoietin-like proteins 1 and 2 in zebrafish vascular development. Proc. Natl. Acad. Sci. USA 2005, 102, 13502–13507. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.S.; Kowanetz, M.; Wu, X.; Zhuang, G.; Ngu, H.; Finkle, D.; Komuves, L.; Peale, F.; Ferrara, N. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J. Pathol. 2012, 227, 404–416. [Google Scholar] [CrossRef]
- Welti, J.C.; Powles, T.; Foo, S.; Gourlaouen, M.; Preece, N.; Foster, J.; Frentzas, S.; Bird, D.; Sharpe, K.; Van Weverwijk, A. Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis 2012, 15, 623–641. [Google Scholar] [CrossRef] [Green Version]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [Green Version]
- Rovida, A.; Castiglioni, V.; Decio, A.; Scarlato, V.; Scanziani, E.; Giavazzi, R.; Cesca, M. Chemotherapy counteracts metastatic dissemination induced by antiangiogenic treatment in mice. Mol. Cancer Ther. 2013, 12, 2237–2247. [Google Scholar] [CrossRef] [Green Version]
- Gaianigo, N.; Melisi, D.; Carbone, C. EMT and Treatment Resistance in Pancreatic Cancer. Cancers 2017, 9, 122. [Google Scholar] [CrossRef]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.-Y.; Fattet, L.; Yang, J. Molecular pathways: Linking tumor microenvironment to epithelial–mesenchymal transition in metastasis. Clin. Cancer Res. 2015, 21, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Padua, D.; Massagué, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Cho, H.J.; Baek, K.E.; Saika, S.; Jeong, M.-J.; Yoo, J. Snail is required for transforming growth factor-β-induced epithelial–mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem. Biophys. Res. Commun. 2007, 353, 337–343. [Google Scholar] [CrossRef]
- Naber, H.P.; Drabsch, Y.; Snaar-Jagalska, B.E.; ten Dijke, P.; van Laar, T. Snail and Slug, key regulators of TGF-β-induced EMT, are sufficient for the induction of single-cell invasion. Biochem. Biophys. Res. Commun. 2013, 435, 58–63. [Google Scholar] [CrossRef]
- Grunewald, M.; Avraham, I.; Dor, Y.; Bachar-Lustig, E.; Itin, A.; Yung, S.; Chimenti, S.; Landsman, L.; Abramovitch, R.; Keshet, E. VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cell 2006, 124, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Crawford, Y.; Ferrara, N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol. Sci. 2009, 30, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed Res. Int. 2013. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Ferrara, N. Refractoriness to antivascular endothelial growth factor treatment: Role of myeloid cells. Cancer Res. 2008, 68, 5501–5504. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+ CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Marigo, I.; Dolcetti, L.; Serafini, P.; Zanovello, P.; Bronte, V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 2008, 222, 162–179. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.-P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+ Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.-F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.-N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Clarke, J.M.; Hurwitz, H.I. Understanding and targeting resistance to anti-angiogenic therapies. J. Gastrointest. Oncol. 2013, 4, 253. [Google Scholar]
- Mahdipour, E.; Mace, K.A. Hox transcription factor regulation of adult bone-marrow-derived cell behaviour during tissue repair and regeneration. Expert Opin. Biol. Ther. 2011, 11, 1079–1090. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Name | Molecular Target | Disease |
|---|---|---|
| Bevacizumab | VEGFA | Recurrent glioblastoma, metastatic colorectal cancer, metastatic non-squamous non-small cell lung, metastatic cervical cancer, metastatic renal cell carcinoma, recurrent epithelial ovarian cancer, fallopian tube cancer |
| Ramucirumab | VEGFR2 | Advanced gastroesophageal junction adenocarcinoma and gastric adenocarcinoma, metastatic colorectal cancer, metastatic non-small cell lung cancer |
| Aflibercept | VEGFA, VEGFB, PIGF | Metastatic colorectal cancer |
| Sorafenib | VEGFRs, PDGFRs | Metastatic thyroid carcinoma, advanced renal cell carcinoma, advanced hepatocellular carcinoma |
| Sunitinib | VEGFRs, PDGFRs | Pancreatic neuroendocrine tumors, metastatic gastrointestinal stromal tumors, advanced renal cell carcinoma |
| Pazopanib | VEGFRs, PDGFRs, FGFRs | Advanced soft tissue carcinoma, advanced renal cell carcinoma |
| Axitinib | VEGFRs, PDGFRs, | Advanced renal cell carcinoma |
| Regorafenib | VEGFRs, PDGFRs, FGFRs | Advanced gastrointestinal stromal tumors, metastatic colorectal cancer, refractory hepatocellular carcinoma |
| Vandetanib | VEGFRs | Metastatic medullary thyroid cancer |
| Cabozantinib | VEGFRs, Tie2 | Metastatic medullary thyroid cancer, refractory advanced renal carcinoma, refractory hepatocellular carcinoma |
| Lenvatinib | VEGFRs, PDGFRs, FGFRs | Recurrent and metastatic thyroid cancer, advanced hepatocellular carcinoma, advanced renal cell carcinoma, advanced endometrial carcinoma |
| Thalidomide | VEGFs, bFGF | Multiple myeloma |
| Lenalidomide | VEGFs, bFGF | Multiple myeloma, myelodysplastic syndromes, mantle cell lymphoma, follicular lymphoma, marginal zone lymphoma |
| Everolimus | mTOR | Advanced renal cell carcinoma, pancreatic neuroendocrine tumors, advanced breast cancer, subependymal giant cell astrocytoma |
| Tumor Type | Molecular Mechanism | Biological Effect | Clinical Observation | Reference |
|---|---|---|---|---|
| Breast cancer | It is the target gene of E2F1 transcription factor. Increased expression of VEGFA, bFGF, IL-8, and Angptl2.Enhanced EMT. | Produces highly vascularized tumors which developed lung metastases. It is involved in the DNA damage response and radiation resistance. | Overexpression is correlated with high tumor grade and poor survival. | [11,50,54,55,56] |
| Colorectal cancer | Increased expression of VEGFA, bFGF TGF-β and IL-8. Enhanced EMT. | Increases cell migration and invasion. The acetylated form decreases cancer progression. | Overexpression is correlated with distal metastasis and resistance to bavacizumab. | [14,49,57,58] |
| Endometrial cancer | Promoted E2F3 expression by direct targeting to its promoter. | Enhances cell migration and cancer progression. | High HOXB9 expression is associated with high histological grade and lymph node metastasis. | [59] |
| Gastric cancer | Suppress the phosphorylation of Akt and NF-κB activity. Induced MET. | Inhibits proliferation and migration of gastric cancer. | Decreased expression and overexpression is correlated with lymph node metastasis and poor survival. | [52,60] |
| Glioma | Activate the TGF-β1/Smad2 signaling. | Enhances cell proliferation, migration and sphere formation and increased tumorigenicity. | Overexpression is correlated with lymph node metastasis and poor survival. | [51] |
| Hepatocellular carcinoma | Enhanced EMT through the TGF-β1/Smad2 signaling. Regulated pro-angiogenic factors. | Promotes cell proliferation, migration, and invasion. | Overexpression is correlated with vascular invasion and poor prognosis. | [13,61,62] |
| Lung cancer | It is target gene of the WNT/TCF4 pathway. GalNAc-T14 induces expression of HOXB9 through Wnt signaling. PCFA-mediated HOXB9 acetylation. | Promotes cell invasion and mediates chemotactic invasion and colony outgrowth. The acetylated form decreases its capacity in promoting cell migration and tumor growth. | Overexpression is correlated with high tumor grade and poor prognosis. | [12,58,63,64] |
| Oral squamous carcinoma | Promoted EMT by TGF-β1/Smad2/Slug signaling. | Enhanced cell migration and invasion. | High HOXB9 levels are associated with high histological grade and shorter overall survival. | [65,66] |
| Ovarian and renal cancer | It is target gene of the miR-192. | Enhanced tumor angiogenesis. | [27] | |
| Pancreatic cancer | Increased expression of VEGFA, bFGF, IL-8 and Angptl2. Enhanced EMT. | Promoted cell proliferation, migration, invasion, and sustained resistance to anti-VEGF inhibition. The acetylated form decreases tumor progression. | Overexpression is associated with shorter overall survival. | [14,67] |
| Prostate cancer | Enhanced EMT Regulated pro-angiogenic factors expression. | Promoted cell proliferation, migration, invasion, and angiogenesis ability. | Overexpression is correlated with vascular invasion and poor prognosis. | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contarelli, S.; Fedele, V.; Melisi, D. HOX Genes Family and Cancer: A Novel Role for Homeobox B9 in the Resistance to Anti-Angiogenic Therapies. Cancers 2020, 12, 3299. https://doi.org/10.3390/cancers12113299
Contarelli S, Fedele V, Melisi D. HOX Genes Family and Cancer: A Novel Role for Homeobox B9 in the Resistance to Anti-Angiogenic Therapies. Cancers. 2020; 12(11):3299. https://doi.org/10.3390/cancers12113299
Chicago/Turabian StyleContarelli, Serena, Vita Fedele, and Davide Melisi. 2020. "HOX Genes Family and Cancer: A Novel Role for Homeobox B9 in the Resistance to Anti-Angiogenic Therapies" Cancers 12, no. 11: 3299. https://doi.org/10.3390/cancers12113299
APA StyleContarelli, S., Fedele, V., & Melisi, D. (2020). HOX Genes Family and Cancer: A Novel Role for Homeobox B9 in the Resistance to Anti-Angiogenic Therapies. Cancers, 12(11), 3299. https://doi.org/10.3390/cancers12113299