Animal Models: A Useful Tool to Unveil Metabolic Changes in Hepatocellular Carcinoma



Abstract
:Simple Summary
Abstract
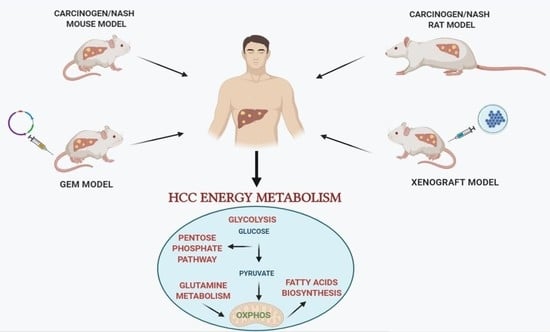
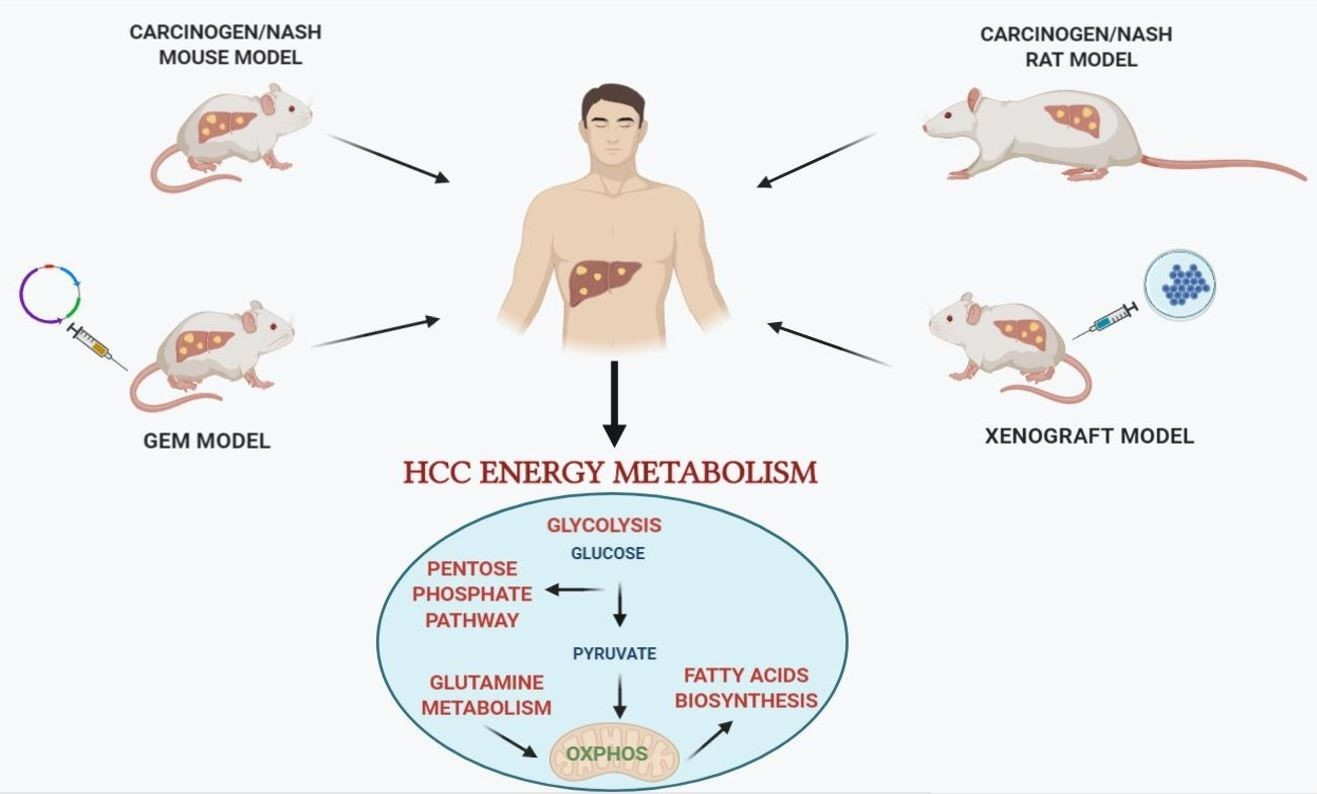{kind=link}
{kind=link}
{kind=link}
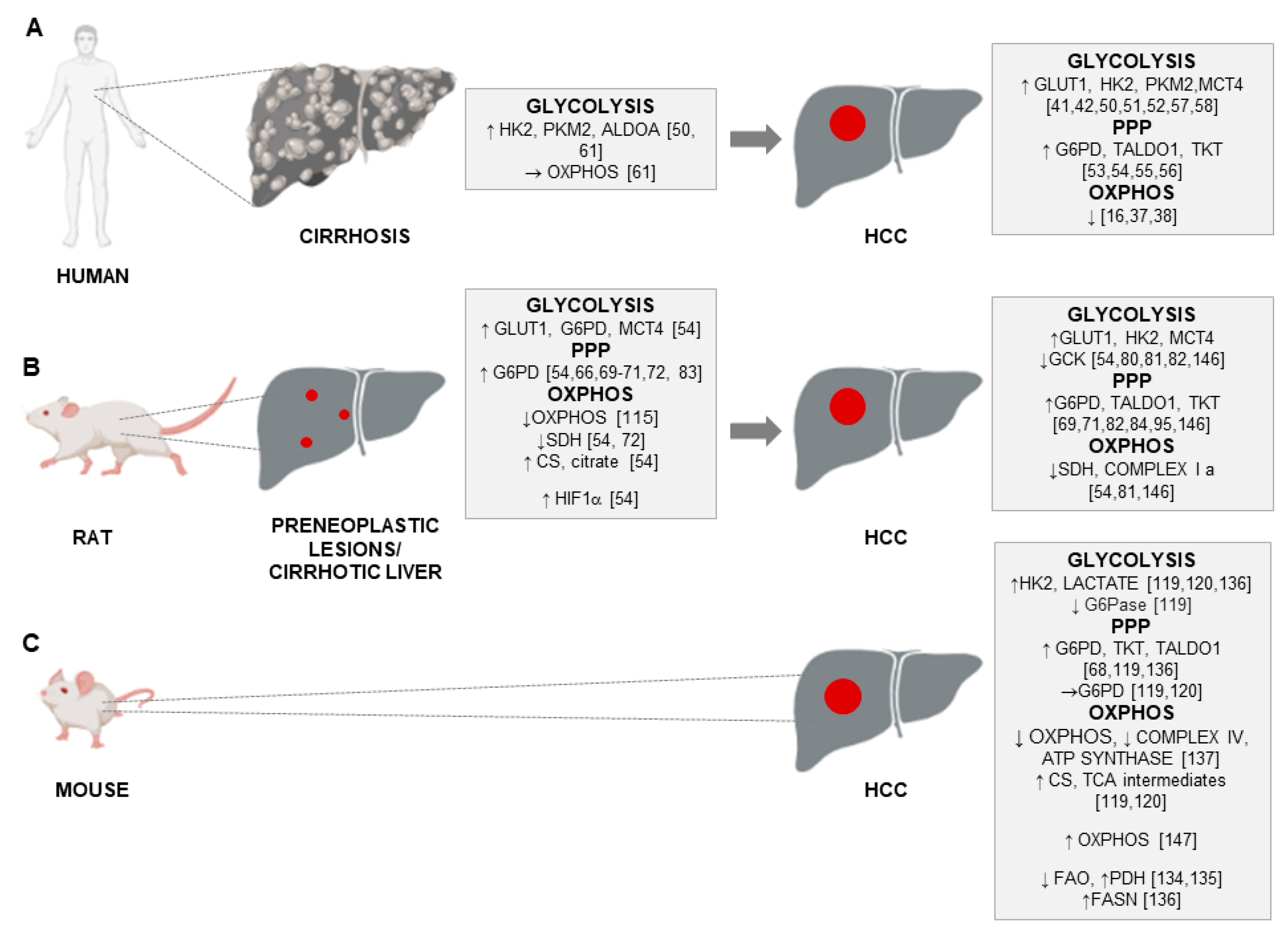{kind=link}
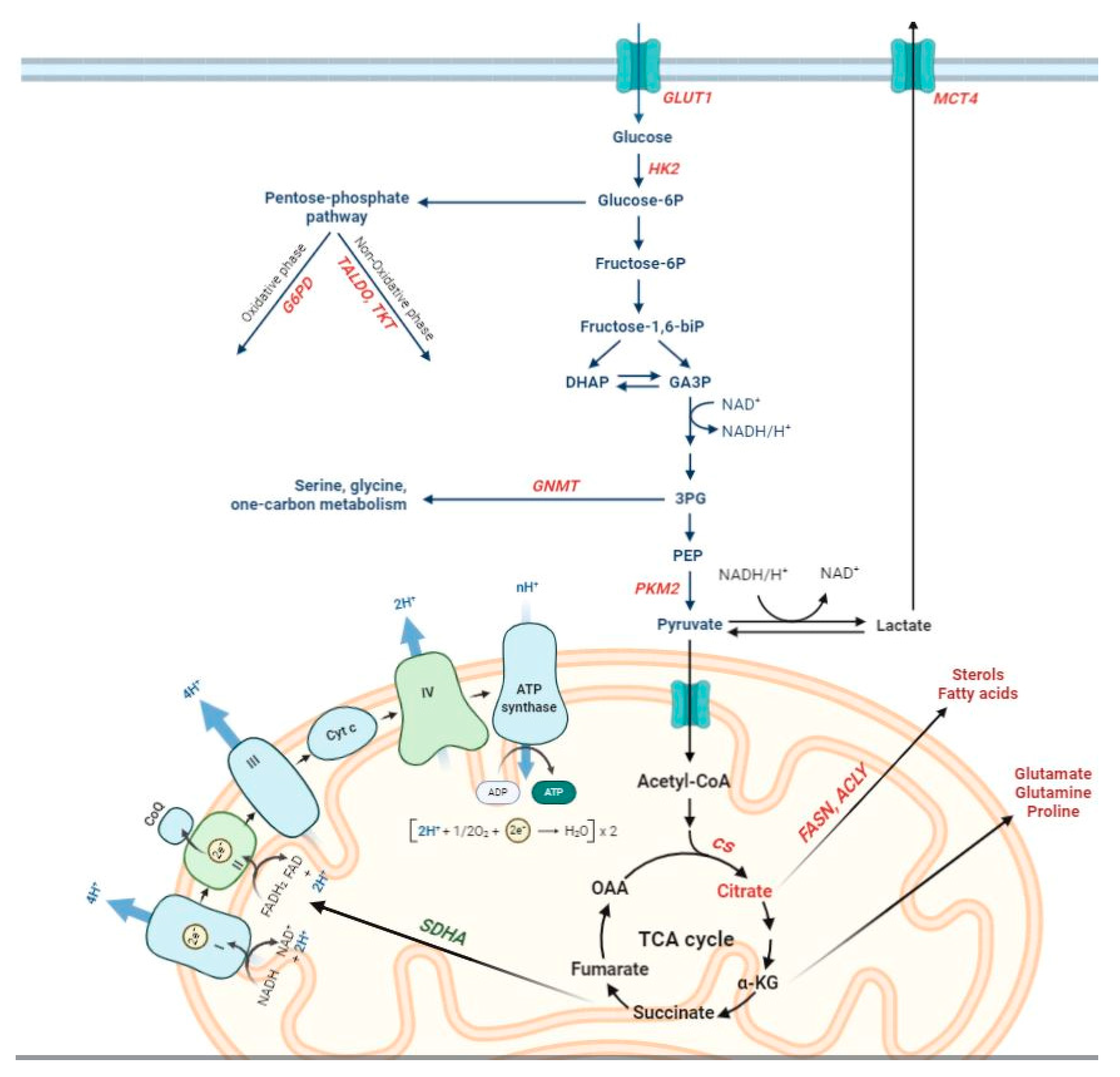{kind=link}
1. Introduction
1.1. Hepatocellular Carcinoma (HCC)
1.2. Metabolic Changes Observed in Human HCC
2. Animal Models of Hepatocarcinogenesis
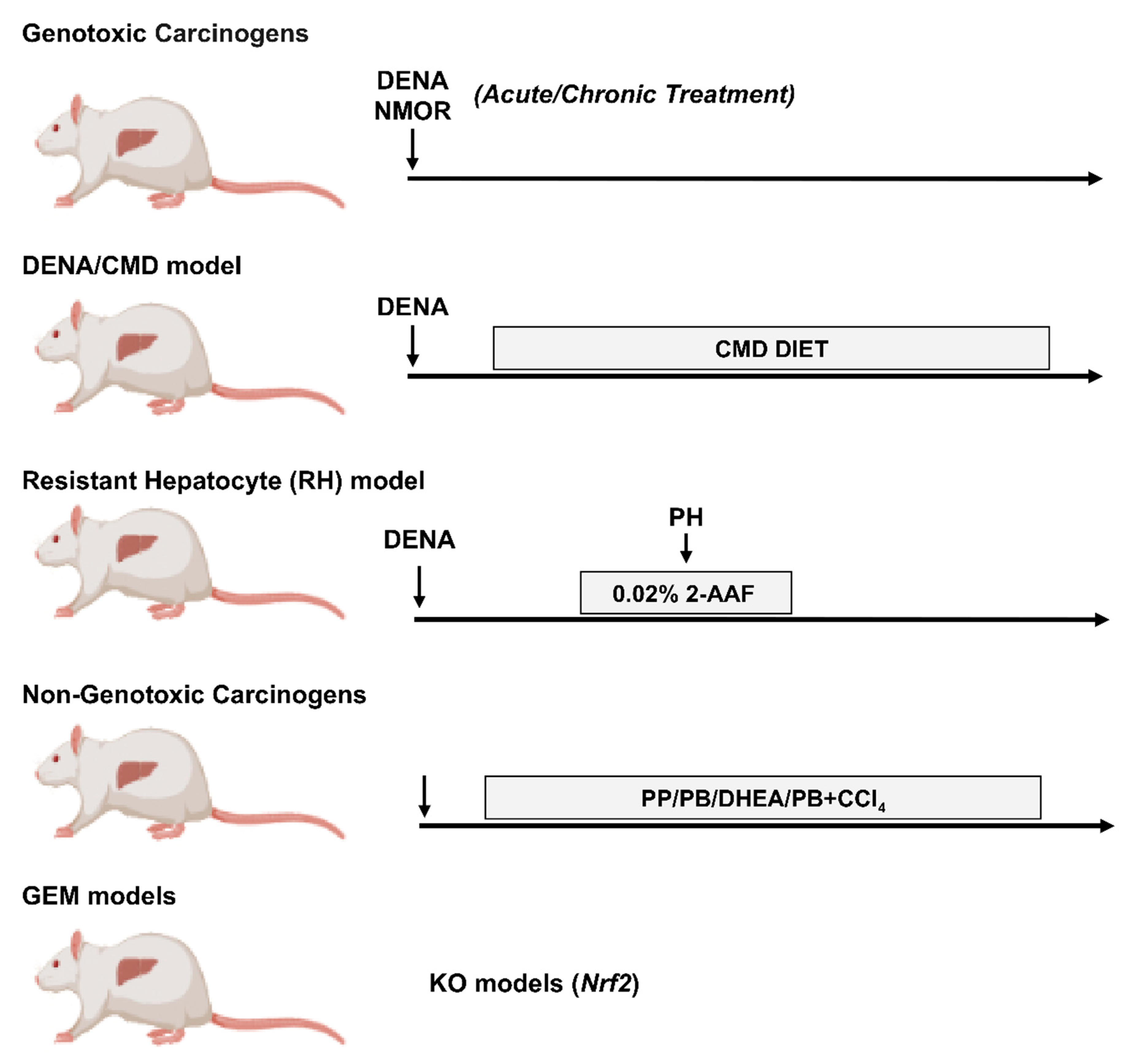
3. Chemically-Induced Models of HCC
4. Rat Models of HCC
4.1. DENA
4.2. DENA/Choline-Deficient, Methionine-Restricted Diet (CMD) Model
4.3. DENA/Resistant Hepatocyte (RH) Model
4.4. Non-Genotoxic Compounds
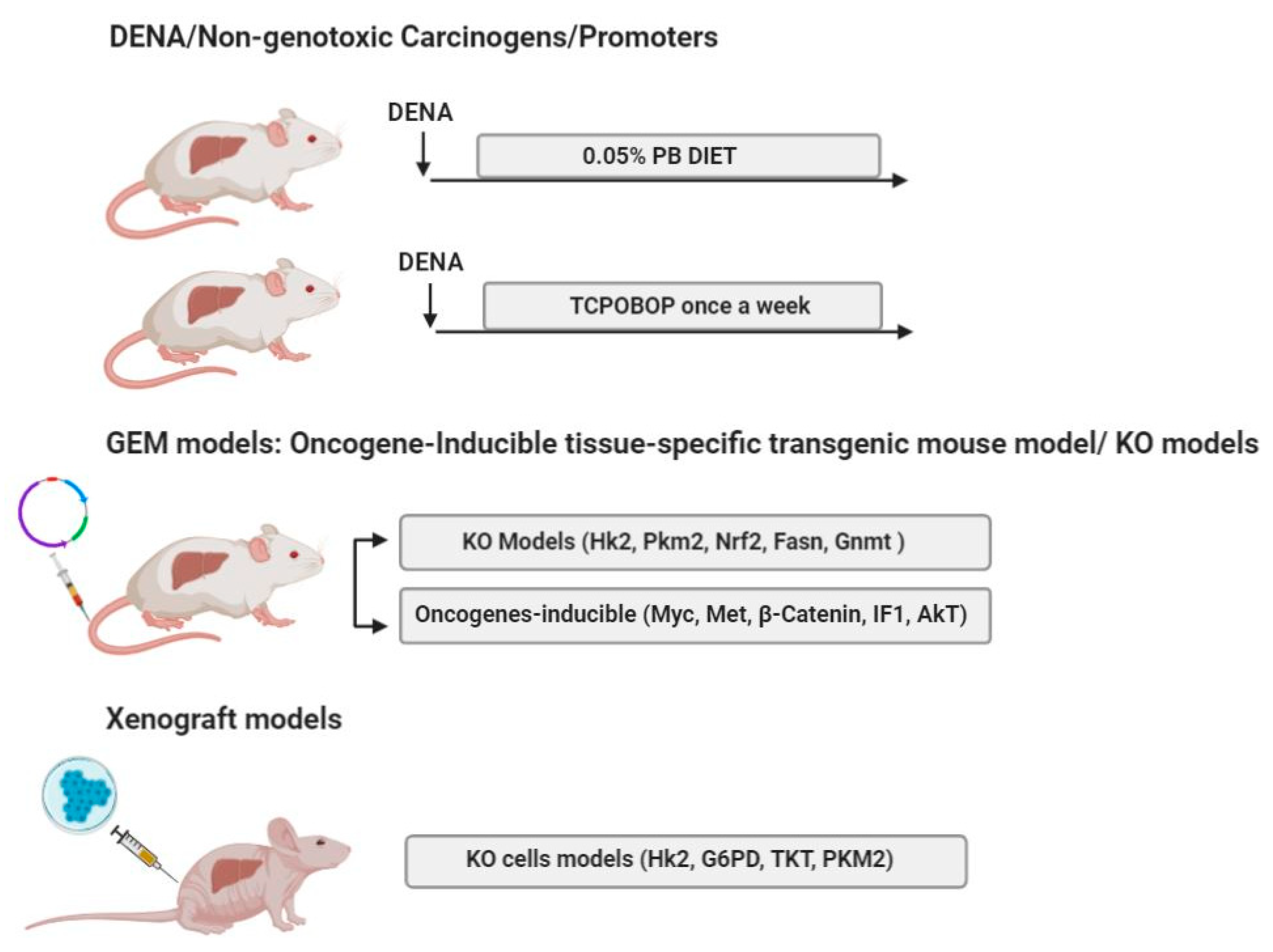
5. Mouse Models of HCC
5.1. DENA/Non-Genotoxic Liver Tumor Carcinogens/Promoters
5.2. GEM Models, Oncogene-Inducible Tissue-Specific Transgenic Mouse Model
5.3. Knockout (KO) Models
5.4. Xenograft Mouse Models
6. MicroRNA and Metabolic Reprogramming
7. Pharmacological Targeting of Warburg Metabolism in HCC
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Finn, R.S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Gerolami, R.; Caparello, C.; et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol. 2018, 69, 353–358. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal. Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Huang, X.Y.; Qiu, S.J.; Yamamoto, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.Z.; Shi, Y.H.; Xiao, Y.S.; et al. Overexpression of PDL-1 significantly associates with tumor aggressiveness and post-operative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef]
- Yarchoan, M.; Xing, D.; Luan, L.; Xu, H.; Sharma, R.B.; Popovic, A.; Pawlik, T.M.; Kim, A.K.; Zhu, Q.; Jaffee, E.M.; et al. Characterization of the Immune Microenvironment in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 7333–7339. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Electronic address: [email protected]; Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Yamamoto, S.; Tatsuno, K.; Renard-Guillet, C.; Tsuji, S.; Hayashi, A.; Ueda, H.; Fukuda, S.; Fujita, T.; Katoh, H.; et al. Accumulation of molecular aberrations distinctive to hepatocellular carcinoma progression. Cancer Res. 2020, 80, 3810–3819. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Mayers, J.R.; Vander Heiden, M.G. Nature and Nurture: What Determines Tumor Metabolic Phenotypes? Cancer Res. 2017, 77, 3131–3134. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer. 2016, 16, 680–693. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, R.Z.; Qu, S.B.; Wang, D.S. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J. Gastroenterol. 2016, 22, 9933–9943. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L. Understanding mouse models of disease through metabolomics. Curr. Opin. Chem. Biol. 2006, 10, 309–315. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Nie, X.; Feng, X.; Chen, W.; Yue, Y.; Tang, H.; Deng, F. Metabonomic studies of human hepatocellular carcinoma using high-resolution magic-angle spinning 1H NMR spectroscopy in conjunction with multivariate data analysis. J. Proteome Res. 2007, 6, 2605–2614. [Google Scholar] [CrossRef]
- Beyoğlu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [Green Version]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323.e1. [Google Scholar] [CrossRef] [Green Version]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.S.; Stoeltzing, O.; Warnecke, C.; Schölmerich, J.; Oefner, P.J.; et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.L.; OuYang, H.Y.; Le, Y.; Jiang, P.; Tang, H.; Yu, Z.S.; He, M.K.; Tang, Y.Q.; Shi, M. Aberrant MCT4 and GLUT1 expression is correlated with early recurrence and poor prognosis of hepatocellular carcinoma after hepatectomy. Cancer Med. 2018, 7, 5339–5350. [Google Scholar] [CrossRef] [PubMed]
- Paudyal, B.; Paudyal, P.; Oriuchi, N.; Tsushima, Y.; Nakajima, T.; Endo, K. Clinical Implications of glucose transport and metabolism evaluated by 18F-FDG PET in hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 1047–1054. [Google Scholar]
- Daskalow, K.; Pfander, D.; Weichert, W.; Rohwer, N.; Thelen, A.; Neuhaus, P.; Jonas, S.; Wiedenmann, B.; Benckert, C.; Cramer, T. Distinct temporospatial expression patterns of glycolysis-related proteins in human hepatocellular carcinoma. Histochem. Cell Biol. 2009, 132, 21–31. [Google Scholar] [CrossRef]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.E.; Kim, J.Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Kim, T.H.; Hyun, D.W.; et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellularcarcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Hao, Y.; Zhou, X.; Li, H.; Liu, F.; Zhu, H.; Song, X.; Niu, Z.; Ni, Q.; Chen, M.S.; et al. Prognostic value of glucose transporter 3 expression in hepatocellular carcinoma. Oncol. Lett. 2020, 19, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Aberrant glycolytic metabolism of cancer cells: A remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J. Bioenerg. Biomembr. 1997, 29, 339–343. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.F.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 2002, 1555, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Guzman, G.; Chennuri, R.; Chan, A.; Rea, B.; Quintana, A.; Patel, R.; Xu, P.Z.; Xie, H.; Hay, N. Evidence for heightened hexokinase II immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig. Dis. Sci. 2015, 60, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Cui, Z.; Chen, P.; Han, H.; Peng, J.; Leng, X. Reduced survival of patients with hepatocellular carcinoma expressing hexokinase II. Med. Oncol. 2012, 29, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Kwee, S.A.; Hernandez, B.; Chan, O.; Wong, L. Choline kinase alpha and hexokinase-2 protein expression in hepatocellular carcinoma: Association with survival. PLoS ONE 2012, 7, e46591. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Song, R.; Song, H.; Zheng, T.; Wang, J.; Liang, Y.; Qi, S.; Lu, Z.; Song, X.; Jiang, H.; et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut 2014, 63, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Ding, X.; Yang, Y.; Zhang, H.; Li, H.; Tong, S.; An, X.; Zhong, Q.; Liu, X.; Ma, L.; et al. Changes in glucose-6-phosphate dehydrogenase expression results in altered behavior of HBV-associated liver cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G611–G622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, I.M.; Lai, R.K.; Lin, S.H.; Tse, A.P.; Chiu, D.K.; Koh, H.Y.; Law, C.T.; Wong, C.M.; Cai, Z.; Wong, C.C.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lu, X.; Wang, Z.; Jin, G.; Wang, Q.; Chen, D.; Chen, T.; Li, J.; Fan, J.; Cong, W.; et al. Co-expression of PKM2 and TRIM35 predicts survival and recurrence in hepatocellular carcinoma. Oncotarget 2015, 6, 2538–2548. [Google Scholar] [CrossRef]
- Wright, T.L. Regenerating nodules—are they premalignant lesions? Hepatology 1991, 13, 1254–1255. [Google Scholar] [CrossRef]
- Pinter, M.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Cancer and liver cirrhosis: Implications on prognosis and management. ESMO Open 2016, 1, e000042. [Google Scholar] [CrossRef]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates with the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 31, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Piorońska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wölfl, S.; Meyer, C.; et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. 2018, 37, 211. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Kisseleva, T.; Francis, H.; Baroni, G.S.; Benedetti, A.; Brenner, D.; Alvaro, D.; Alpini, G.; Marzioni, M. Liver carcinogenesis: Rodent models of hepatocarcinoma and cholangiocarcinoma. Dig. Liver Dis. 2013, 45, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, N.P.; Colaço, A.A.; Oliveira, P.A. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Vesselinovitch, S.D.; Mihailovich, N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983, 43, 4253–4259. [Google Scholar]
- Klimek, F.; Mayer, D.; Bannasch, P. Biochemical microanalysis of glycogen content and glucose-6-phosphate dehydrogenase activity in focal lesions of the rat liver induced by N-nitrosomorpholine. Carcinogenesis 1984, 5, 265–268. [Google Scholar] [CrossRef]
- Bannasch, P. Pathobiology of chemical hepatocarcinogenesis: Recent progress and perspectives. Part II. Metabolic and molecular changes. J. Gastroenterol. Hepatol. 1990, 5, 310–320. [Google Scholar] [CrossRef]
- Hacker, H.J.; Mtiro, H.; Bannasch, P.; Vesselinovitch, S.D. Histochemical profile of mouse hepatocellular adenomas and carcinomas induced by a single dose of diethylnitrosamine. Cancer Res. 1991, 51, 1952–1958. [Google Scholar]
- Baba, M.; Yamamoto, R.; Iishi, H.; Tatsuta, M.; Wada, A. Role of glucose-6-phosphate dehydrogenase on enhanced proliferation of pre-neoplastic and neoplastic cells in rat liver induced by N-nitrosomorpholine. Int. J. Cancer 1989, 43, 892–895. [Google Scholar] [CrossRef]
- Cortinovis, C.; Klimek, F.; Nogueira, E. Rat hepatocarcinogenesis induced by N-nitrosodiethylamine and N-nitrosomorpholine continuously administered at low doses. From basophilic areas of hepatocytes to hepatocellular tumors. Am. J. Pathol. 1991, 139, 1157–1171. [Google Scholar]
- Stumpf, H.; Bannasch, P. Overexpression of glucose-6-phosphate-dehydrogenase in rat hepatic preneoplasia and neoplasia. Int. J. Oncol. 1994, 5, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.A.; Tsuda, H.; Ito, N. Dehydrogenase histochemistry of N-ethyl-N- hydroxyethylnitrosamine-induced focal liver lesions in the rat—increase in NADPH-generating capacity. Carcinogenesis 1986, 7, 339–342. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.J. Interaction of chemical carcinogens with macromolecules. J. Cancer Res. Clin. Oncol. 1981, 99, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Cameron, R.; Farber, E. Some conclusions derived from a liver model for carcinogenesis. Natl. Cancer Inst. Monogr. 1981, 58, 49–53. [Google Scholar]
- Tsuda, H.; Farber, E. Resistant hepatocytes as early changes in liver induced by polycyclic aromatic hydrocarbons. Int. J. Cancer. 1980, 25, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Magee, P.N.; Lee, K.Y. Cellular injury and carcinogenesis. Alkylation of ribonucleic acid of rat liver by diethylnitrosamine and n-butylmethylnitrosamine in vivo. Biochem. J. 1964, 91, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Verna, L.; Whysner, J.; Williams, G.M. N-nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Hacker, H.J.; Moore, M.A.; Mayer, D.; Bannasch, P. Correlative histochemistry of some enzymes of carbohydrate metabolism in preneoplastic and neoplastic lesions in the rat liver. Carcinogenesis 1982, 3, 1265–1272. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.F.; Wang, J.; Huang, C.; Zhang, S.; Yang, J.; Jiang, A.; Zhou, R.; Pan, D. Gas chromatography/time-of-flight mass spectrometry-based metabonomics of hepatocarcinoma in rats with lung metastasis: Elucidation of the metabolic characteristics of hepatocarcinoma at formation and metastasis. Rapid Commun. Mass Spectrom. 2010, 24, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, S.; Li, Z.; Yang, J.; Huang, C.; Liang, R.; Liu, Z.; Zhou, R. (1)H-NMR-based metabolomics of tumor tissue for the metabolic characterization of rat hepatocellular carcinoma formation and metastasis. Tumour Biol. 2011, 32, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Frederiks, W.M.; Vizan, P.; Bosch, K.S.; Vreeling-Sindelárová, H.; Boren, J.; Cascante, M. Elevated activity of the oxidative and non-oxidative pentose phosphate pathway in (pre)neoplastic lesions in rat liver. Int. J. Exp. Pathol. 2008, 89, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Orrù, C.; Szydlowska, M.; Taguchi, K.; Zavattari, P.; Perra, A.; Yamamoto, M.; Columbano, A. Genetic inactivation of Nrf2 prevents clonal expansion of initiated cells in a nutritional model of rat hepatocarcinogenesis. J. Hepatol. 2018, 69, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, B.; Pani, P.; Schlunk, F.F. Choline-deficiency fatty liver: Impaired release of hepatic triglycerides. J. Lipid Res. 1968, 9, 437–446. [Google Scholar] [PubMed]
- Koteish, A.; Mae Diehl, A. Animal models of steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Shinozuka, H.; Sells, M.A.; Katyal, S.L.; Sell, S.; Lombardi, B. Effects of a choline-devoid diet on the emergence of gamma-glutamyltranspeptidase-positive foci in the liver of carcinogen-treated rats. Cancer Res. 1979, 39, 2515–2521. [Google Scholar]
- Yokoyama, S.; Sells, M.A.; Reddy, T.V.; Lombardi, B. Hepatocarcinogenic and promoting action of a choline-devoid diet in the rat. Cancer Res. 1985, 45, 2834–2842. [Google Scholar]
- Sato, K.; Satoh, K.; Tsuchida, S.; Hatayama, I.; Shen, H.; Yokoyama, Y.; Yamada, Y.; Tamai, K. Specific expression of glutathione S-transferase Pi forms in (pre)neoplastic tissues: Their properties and functions. Tohoku J. Exp. Med. 1992, 168, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 26, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, M.; Sordelli, I.M.; Lombardi, A.; Lamberti, M.; Tarantino, L.; Giudice, A.; Stiuso, P.; Abbruzzese, A.; Sperlongano, R.; Accardo, M.; et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: An overview. J. Transl. Med. 2011, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Chung, F.L. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Orrù, C.; Perra, A.; Kowalik, M.A.; Rizzolio, S.; Puliga, E.; Cabras, L.; Giordano, S.; Columbano, A. Distinct Mechanisms Are Responsible for Nrf2-Keap1 Pathway Activation at Different Stages of Rat Hepatocarcinogenesis. Cancers (Basel) 2020, 12, 2305. [Google Scholar] [CrossRef]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 1977, 88, 595–618. [Google Scholar]
- Kowalik, M.A.; Sulas, P.; Ledda-Columbano, G.M.; Giordano, S.; Columbano, A.; Perra, A. Cytokeratin-19 positivity is acquired along cancer progression and does not predict cell origin in rat hepatocarcinogenesis. Oncotarget 2015, 6, 38749–38763. [Google Scholar] [CrossRef]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef]
- Yang, X.R.; Xu, Y.; Yu, B.; Zhou, J.; Qiu, S.J.; Shi, G.M.; Zhang, B.H.; Wu, W.Z.; Shi, Y.H.; Wu, B.; et al. High expression levels of putative hepatic stem/progenitor cell biomarkers related to tumour angiogenesis and poor prognosis of hepatocellular carcinoma. Gut 2010, 59, 953–962. [Google Scholar] [CrossRef]
- Andersen, J.B.; Loi, R.; Perra, A.; Factor, V.M.; Ledda-Columbano, G.M.; Columbano, A.; Thorgeirsson, S.S. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology 2010, 51, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014, 24, 455–463. [Google Scholar]
- Guzzo, G.; Sciacovelli, M.; Bernardi, P.; Rasola, A. Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget 2014, 5, 11897–11908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y. Overview of genotoxic carcinogens and non-genotoxic carcinogens. Exp. Toxicol. Pathol. 1992, 44, 465–471. [Google Scholar] [CrossRef]
- Williams, G.M. Chemicals with carcinogenic activity in the rodent liver; mechanistic evaluation of human risk. Cancer Lett. 1997, 117, 175–188. [Google Scholar] [CrossRef]
- Lee, S.J.; Yum, Y.N.; Kim, S.C.; Kim, Y.; Lim, J.; Lee, W.J.; Koo, K.H.; Kim, J.H.; Kim, J.E.; Lee, W.S.; et al. Distinguishing between genotoxic and non-genotoxic hepatocarcinogens by gene expression profiling and bioinformatic pathway analysis. Sci. Rep. 2013, 3, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwan, B.A.; Lubet, R.A.; Ward, J.M.; Hrabie, J.A.; Rice, J.M. Tumor-promoting and hepatocarcinogenic effects of 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) in DBA/2NCr and C57BL/6NCr mice and an apparent promoting effect on nasal cavity tumors but not on hepatocellular tumors in F344/NCr rats initiated with N-nitrosodiethylamine. Carcinogenesis 1992, 13, 1893–1901. [Google Scholar] [PubMed]
- Tzameli, I.; Pissios, P.; Schuetz, E.G.; Moore, D.D. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell Biol. 2000, 20, 2951–2958. [Google Scholar] [PubMed] [Green Version]
- Rao, M.S.; Tatematsu, M.; Subbarao, V.; Ito, N.; Reddy, J.K. Analysis of peroxisome proliferator-induced preneoplastic and neoplastic lesions of rat liver for placental form of glutathione S-transferase and gamma-glutamyltranspeptidase. Cancer Res. 1986, 46, 5287–5290. [Google Scholar] [PubMed]
- Rao, M.S.; Subbarao, V.; Kumar, S.; Yeldandi, A.V.; Reddy, J.K. Phenotypic properties of liver tumors induced by dehydroepiandrosterone in F-344 rats. Jpn. J. Cancer Res. 1992, 83, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Irisarri, E.; Monro, A.M. Hepatic foci of cellular and enzymatic alteration and nodules in rats treated with clofibrate or diethylnitrosamine followed by phenobarbital: Their rate of onset and their reversibility. J. Natl. Cancer Inst. 1986, 76, 475–484. [Google Scholar] [PubMed]
- Rao, K.N.; Elm, M.S.; Kelly, R.H.; Chandar, N.; Brady, E.P.; Rao, B.; Shinozuka, H.; Eagon, P.K. Hepatic hyperplasia and cancer in rats: Metabolic alterations associated with cell growth. Gastroenterology 1997, 113, 238–248. [Google Scholar] [CrossRef]
- Nishikawa, T.; Bellance, N.; Damm, A.; Bing, H.; Zhu, Z.; Handa, K.; Yovchev, M.I.; Sehgal, V.; Moss, T.J.; Oertel, M.; et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J. Hepatol. 2014, 60, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Aydinlik, H.; Nguyen, T.D.; Moennikes, O.; Buchmann, A.; Schwarz, M. Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of beta-catenin-mutated mouse liver tumors. Oncogene 2001, 20, 7812–7816. [Google Scholar] [CrossRef] [Green Version]
- Chafey, P.; Finzi, L.; Boisgard, R.; Caüzac, M.; Clary, G.; Broussard, C.; Pégorier, J.P.; Guillonneau, F.; Mayeux, P.; Camoin, L.; et al. Proteomicanalysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 2009, 9, 3889–3900. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [Green Version]
- Unterberger, E.B.; Eichner, J.; Wrzodek, C.; Lempiäinen, H.; Luisier, R.; Terranova, R.; Metzger, U.; Plummer, S.; Knorpp, T.; Braeuning, A.; et al. Ha-ras and β-catenin oncoproteins orchestrate metabolic programs in mouse liver tumors. Int. J. Cancer. 2014, 135, 1574–1585. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Tward, A.D.; Jones, K.D.; Yant, S.; Cheung, S.T.; Fan, S.T.; Chen, X.; Kay, M.A.; Wang, R.; Bishop, J.M. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc. Natl. Acad. Sci. USA 2007, 104, 14771–14776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattu, S.; Saliba, C.; Sulas, P.; Zavattari, P.; Perra, A.; Kowalik, M.A.; Monga, S.P.; Columbano, A. High Frequency of β-Catenin Mutations in Mouse Hepatocellular Carcinomas Induced by a Nongenotoxic Constitutive Androstane Receptor Agonist. Am. J. Pathol. 2018, 88, 2497–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, C.; Li, P.; Wu, N.L.; Yan, Y.; Ying, Q.L. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 2010, 467, 211–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Takaku, M.; Egner, P.A.; Morita, M.; Kaneko, T.; Mashimo, T.; Kensler, T.W.; Yamamoto, M. Generation of a New Model Rat: Nrf2 Knockout Rats Are Sensitive to Aflatoxin B1 Toxicity. Toxicol. Sci. 2016, 152, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M. Finding an “Achilles’ heel” of cancer: The role of glucose and glutamine metabolism in the survival of transformed cells. Cell Cycle 2008, 7, 2083–2089. [Google Scholar] [CrossRef] [Green Version]
- Fafalios, A.; Ma, J.; Tan, X.; Stoops, J.; Luo, J.; Defrances, M.C.; Zarnegar, R. A hepatocyte growth factor receptor (Met)-insulin receptor hybrid governs hepatic glucose metabolism. Nat. Med. 2011, 17, 1577–1584. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell. 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Monga, S.P. β-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310. [Google Scholar] [CrossRef] [Green Version]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 2001, 153, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxid Med. Cell Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lu, J.; Edmunds, L.R.; Kulkarni, S.; Dolezal, J.; Tao, J.; Ranganathan, S.; Jackson, L.; Fromherz, M.; Beer-Stolz, D.; et al. Coordinated Activities of Multiple Myc-dependent and Myc-independent Biosynthetic Pathways in Hepatoblastoma. J. Biol. Chem. 2016, 291, 26241–26251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolezal, J.M.; Wang, H.; Kulkarni, S.; Jackson, L.; Lu, J.; Ranganathan, S.; Goetzman, E.S.; Bharathi, S.S.; Beezhold, K.; Byersdorfer, C.A.; et al. Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J. Biol. Chem. 2017, 292, 10068–10086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Antonucci, L.; Yamachika, S.; Zhang, Z.; Taniguchi, K.; Umemura, A.; Hatzivassiliou, G.; Roose-Girma, M.; Reina-Campos, M.; Duran, A.; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J. Hepatol. 2020, 72, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santacatterina, F.; Sánchez-Cenizo, L.; Formentini, L.; Mobasher, M.A.; Casas, E.; Rueda, C.B.; Martínez-Reyes, I.; Núñez de Arenas, C.; García-Bermúdez, J.; Zapata, J.M.; et al. Down-regulation of oxidative phosphorylation in the liver by expression of the ATPase inhibitory factor 1 induces a tumor-promoter metabolic state. Oncotarget 2016, 7, 490–508. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Aragó, M.; Formentini, L.; Cuezva, J.M. Mitochondria-mediated energy adaption in cancer: The H(+)-ATP synthase-geared switch of metabolism in human tumors. Antioxid. Redox Signal. 2013, 19, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [Green Version]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Gocheva, V.; Miller, K.M.; Israelsen, W.J.; Bhutkar, A.; Clish, C.B.; Davidson, S.M.; Luengo, A.; Bronson, R.T.; Jacks, T.; et al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016, 30, 1020–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Lucas, A.; Li, X.; Hu, J.; Che, L.; Song, X.; Jia, J.; Wang, J.; Xie, C.; Driscoll, P.C.; Tschaharganeh, D.F.; et al. Glucose Catabolism in Liver Tumors Induced by c-MYC Can Be Sustained by Various PKM1/PKM2 Ratios and Pyruvate Kinase Activities. Cancer Res. 2017, 77, 4355–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef] [Green Version]
- Kowalik, M.A.; Puliga, E.; Cabras, L.; Sulas, P.; Petrelli, A.; Perra, A.; Ledda-Columbano, G.M.; Morandi, A.; Merlin, S.; Orrù, C.; et al. Thyroid hormone inhibits hepatocellular carcinoma progression via induction of differentiation and metabolic reprogramming. J. Hepatol. 2020, 72, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Sugimoto, M.; Arias, E.; Kasashima, H.; Cordes, T.; Linares, J.F.; Duran, A.; Nakanishi, Y.; Nakanishi, N.; L’Hermitte, A.; et al. PKCλ/ι Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 2020, 38, 247–262.e11. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Simile, M.M.; Ribback, S.; Dombrowski, F.; Evert, M.; Chen, X.; Calvisi, D.F. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle 2017, 16, 499–507. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 20484. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Chantar, M.L.; Vázquez-Chantada, M.; Ariz, U.; Martínez, N.; Varela, M.; Luka, Z.; Capdevila, A.; Rodríguez, J.; Aransay, A.M.; Matthiesen, R.; et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 2008, 47, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell, P.; Villanueva, A.; Friedman, S.L.; Koike, K.; Llovet, J.M. Experimental models of hepatocellular carcinoma. J. Hepatol. 2008, 48, 858–879. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef]
- Fornari, F.; Gramantieri, L.; Callegari, E.; Shankaraiah, R.C.; Piscaglia, F.; Negrini, M.; Giovannini, C. MicroRNAs in Animal Models of HCC. Cancers (Basel) 2019, 11, 1906. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Pedroza-Torres, A.; Romero-Córdoba, S.L.; Justo-Garrido, M.; Salido-Guadarrama, I.; Rodríguez-Bautista, R.; Montaño, S.; Muñiz-Mendoza, R.; Arriaga-Canon, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. MicroRNAs in Tumor Cell Metabolism: Roles and Therapeutic Opportunities. Front. Oncol. 2019, 9, 1404. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Qiu, Z.; Wang, Z.; Wang, Q.; Tan, N.; Chen, T.; Chen, Z.; Huang, S.; Gu, J.; Li, J.; et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology 2015, 62, 1132–1144. [Google Scholar] [CrossRef]
- Xu, F.; Yan, J.J.; Gan, Y.; Chang, Y.; Wang, H.L.; He, X.X.; Zhao, Q. miR-885-5p Negatively Regulates Warburg Effect by Silencing Hexokinase 2 in Liver Cancer. Mol. Ther. Nucleic Acids. 2019, 18, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, W.; Li, M.; Hu, Y.; Zhang, H.; Song, G.; Yang, L.; Cai, K.; Luo, Z. Targeted inhibition of MCT4 disrupts intracellular pH homeostasis and confers self- regulated apoptosis on hepatocellular carcinoma. Exp. Cell Res. 2019, 384, 111591. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.A. Biographical sketch: Otto Heinrich Warburg, PhD, MD. Clin. Orthop. Relat. Res. 2010, 468, 2831–2832. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Calvisi, D.F.; Frau, M.; Tomasi, M.L.; Feo, F.; Pascale, R.M. Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: Novel insights from interspecies comparison. Biochim. Biophys. Acta 2012, 1826, 215–237. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Yoon, J.H.; Jeong, J.M.; Cheon, G.J.; Lee, T.S.; Yang, J.I.; Park, S.C.; Lee, H.S. Apoptosis-inducing antitumor efficacy of hexokinase II inhibitor in hepatocellular carcinoma. Mol. Cancer Ther. 2007, 6, 2554–2562. [Google Scholar] [CrossRef] [Green Version]
- Pernicova, I.; Korbonits, M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Wang, H.; Lu, J.; Dolezal, J.; Kulkarni, S.; Zhang, W.; Chen, A.; Gorka, J.; Mandel, J.A.; Prochownik, E.V. Inhibition of hepatocellular carcinoma by metabolic normalization. PLoS ONE 2019, 14, e0218186. [Google Scholar] [CrossRef] [Green Version]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Lévy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebayo Michael, A.O.; Ko, S.; Tao, J.; Moghe, A.; Yang, H.; Xu, M.; Russell, J.O.; Pradhan-Sundd, T.; Liu, S.; Singh, S.; et al. Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by β-Catenin Mutations. Cell Metab. 2019, 29, 1135–1150.e6. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Columbano, A.; Chiellini, G.; Kowalik, M.A. GC-1: A Thyromimetic With Multiple Therapeutic Applications in Liver Disease. Gene Expr. 2017, 17, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puliga, E.; Min, Q.; Tao, J.; Zhang, R.; Pradhan-Sundd, T.; Poddar, M.; Singh, S.; Columbano, A.; Yu, J.; Monga, S.P. Thyroid Hormone Receptor-β Agonist GC-1 Inhibits Met-β-Catenin-Driven Hepatocellular Cancer. Am. J. Pathol. 2017, 187, 2473–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brent, G.A. The molecular basis of thyroid hormone action. N. Engl. J. Med. 1994, 331, 847–853. [Google Scholar] [PubMed]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [Green Version]
- Weitzel, J.M.; Iwen, K.A. Coordination of mitochondrial biogenesis by thyroid hormone. Mol. Cell Endocrinol. 2011, 342, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lazar, M.A. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr. Rev. 1993, 14, 184–193. [Google Scholar]
- Frau, C.; Loi, R.; Petrelli, A.; Perra, A.; Menegon, S.; Kowalik, M.A.; Pinna, S.; Leoni, V.P.; Fornari, F.; Gramantieri, L.; et al. Local hypothyroidism favors the progression of preneoplastic lesions to hepatocellular carcinoma in rats. Hepatology 2015, 61, 249–259. [Google Scholar] [CrossRef]
- Ledda-Columbano, G.M.; Perra, A.; Loi, R.; Shinozuka, H.; Columbano, A. Cell proliferation induced by triiodothyronine in rat liver is associated with nodule regression and reduction of hepatocellular carcinomas. Cancer Res. 2000, 60, 603–609. [Google Scholar] [PubMed]
- Chi, H.C.; Chen, S.L.; Tsai, C.Y.; Chuang, W.Y.; Huang, Y.H.; Tsai, M.M.; Wu, S.M.; Sun, C.P.; Yeh, C.T.; Lin, K.H. Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy 2016, 12, 2271–2285. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene 2017, 36, 5274–5284. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Senese, R.; Lanni, A.; Goglia, F. Thyroid hormones and mitochondria: With a brief look at derivatives and analogues. Mol. Cell Endocrinol. 2013, 379, 51–61. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef] [Green Version]
- Klein, I.; Ojamaa, K. Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef]
- Marks, P.A.; Banks, J. Inhibition of mammalian glucose-6-phosphate dehydrogenase by steroids. Proc. Natl. Acad. Sci. USA 1960, 46, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Garcea, R.; Daino, L.; Pascale, R.; Frassetto, S.; Cozzolino, P.; Ruggiu, M.E.; Feo, F. Inhibition by dehydroepiandrosterone of liver preneoplastic foci formation in rats after initiation-selection in experimental carcinogenesis. Toxicol. Pathol. 1987, 15, 164–169. [Google Scholar] [CrossRef]
- Simile, M.; Pascale, R.M.; De Miglio, M.R.; Nufris, A.; Daino, L.; Seddaiu, M.A.; Muroni, M.R.; Rao, K.N.; Feo, F. Inhibition by dehydroepiandrosterone of growth and progression of persistent liver nodules in experimental rat liver carcinogenesis. Int. J. Cancer 1995, 62, 210–215. [Google Scholar] [CrossRef]
- Ghergurovich, J.M.; García-Cañaveras, J.C.; Wang, J.; Schmidt, E.; Zhang, Z.; TeSlaa, T.; Patel, H.; Chen, L.; Britt, E.C.; Piqueras-Nebot, M.; et al. A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat. Chem. Biol. 2020, 16, 731–739. [Google Scholar] [CrossRef]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef] [PubMed]

 Hydrodynamic Injection.
Hydrodynamic Injection.  Cancer cells. The figure has been prepared by adapting BioRender images.
Hydrodynamic Injection. Cancer cells. The figure has been prepared by adapting BioRender images.
Cancer cells. The figure has been prepared by adapting BioRender images.
Hydrodynamic Injection. Cancer cells. The figure has been prepared by adapting BioRender images.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, M.; Columbano, A.; Perra, A.; Kowalik, M.A. Animal Models: A Useful Tool to Unveil Metabolic Changes in Hepatocellular Carcinoma. Cancers 2020, 12, 3318. https://doi.org/10.3390/cancers12113318
Serra M, Columbano A, Perra A, Kowalik MA. Animal Models: A Useful Tool to Unveil Metabolic Changes in Hepatocellular Carcinoma. Cancers. 2020; 12(11):3318. https://doi.org/10.3390/cancers12113318
Chicago/Turabian StyleSerra, Marina, Amedeo Columbano, Andrea Perra, and Marta Anna Kowalik. 2020. "Animal Models: A Useful Tool to Unveil Metabolic Changes in Hepatocellular Carcinoma" Cancers 12, no. 11: 3318. https://doi.org/10.3390/cancers12113318
APA StyleSerra, M., Columbano, A., Perra, A., & Kowalik, M. A. (2020). Animal Models: A Useful Tool to Unveil Metabolic Changes in Hepatocellular Carcinoma. Cancers, 12(11), 3318. https://doi.org/10.3390/cancers12113318