Cancer Stem Cells and the Slow Cycling Phenotype: How to Cut the Gordian Knot Driving Resistance to Therapy in Melanoma



Abstract
:Simple Summary
Abstract
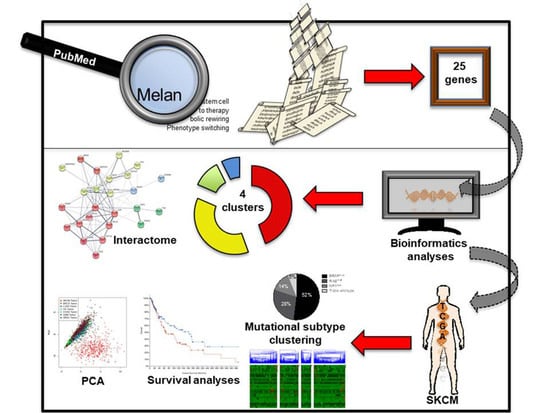
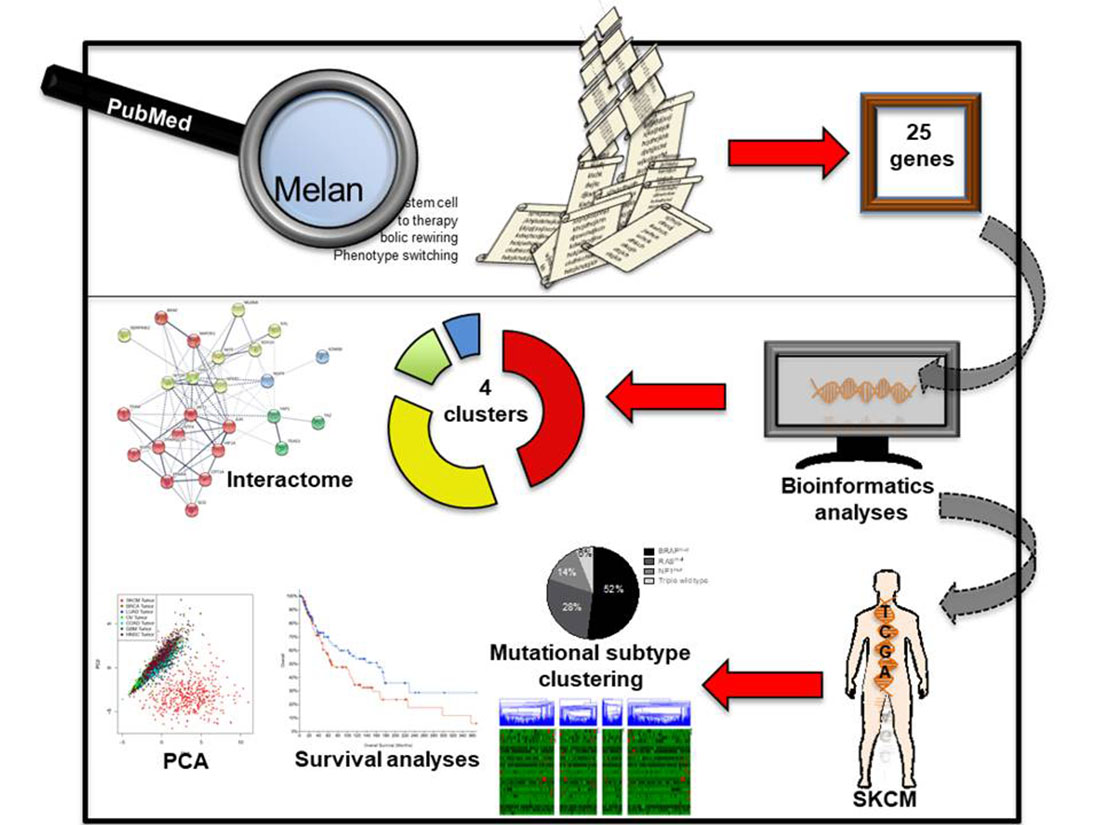
1. Introduction
2. Results
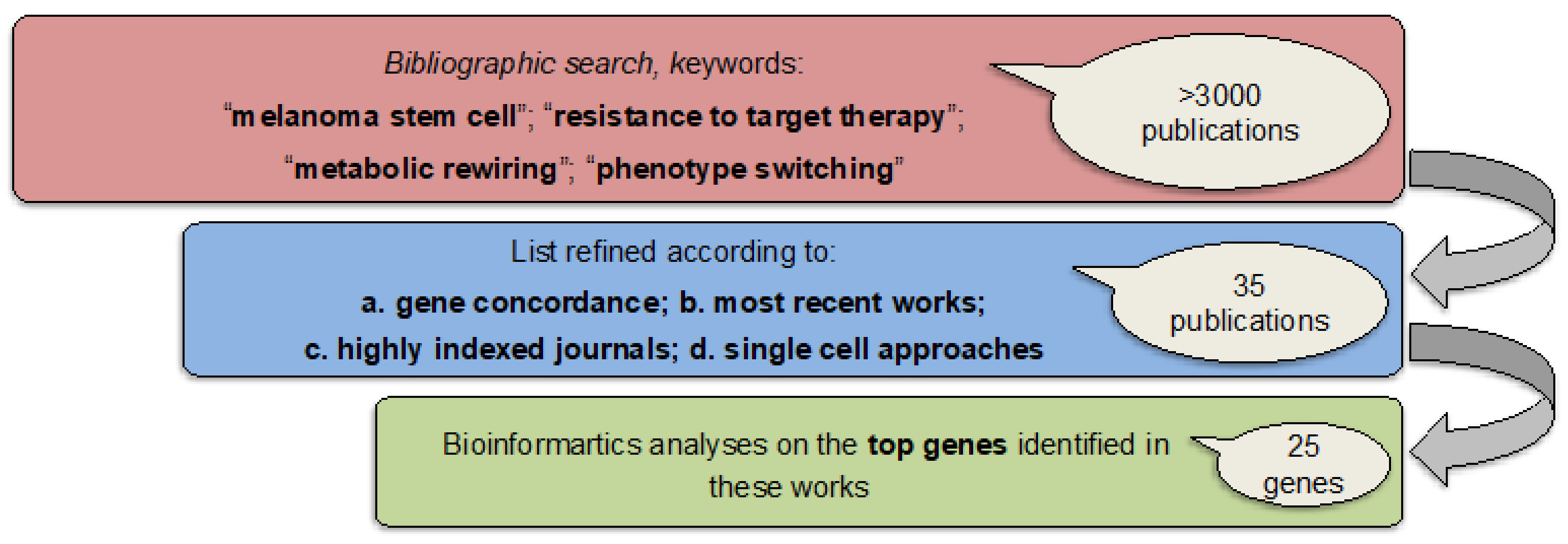
2.1. Identifying Common Denominators for Melanoma Stem Cell Survival through Bibliographic Search
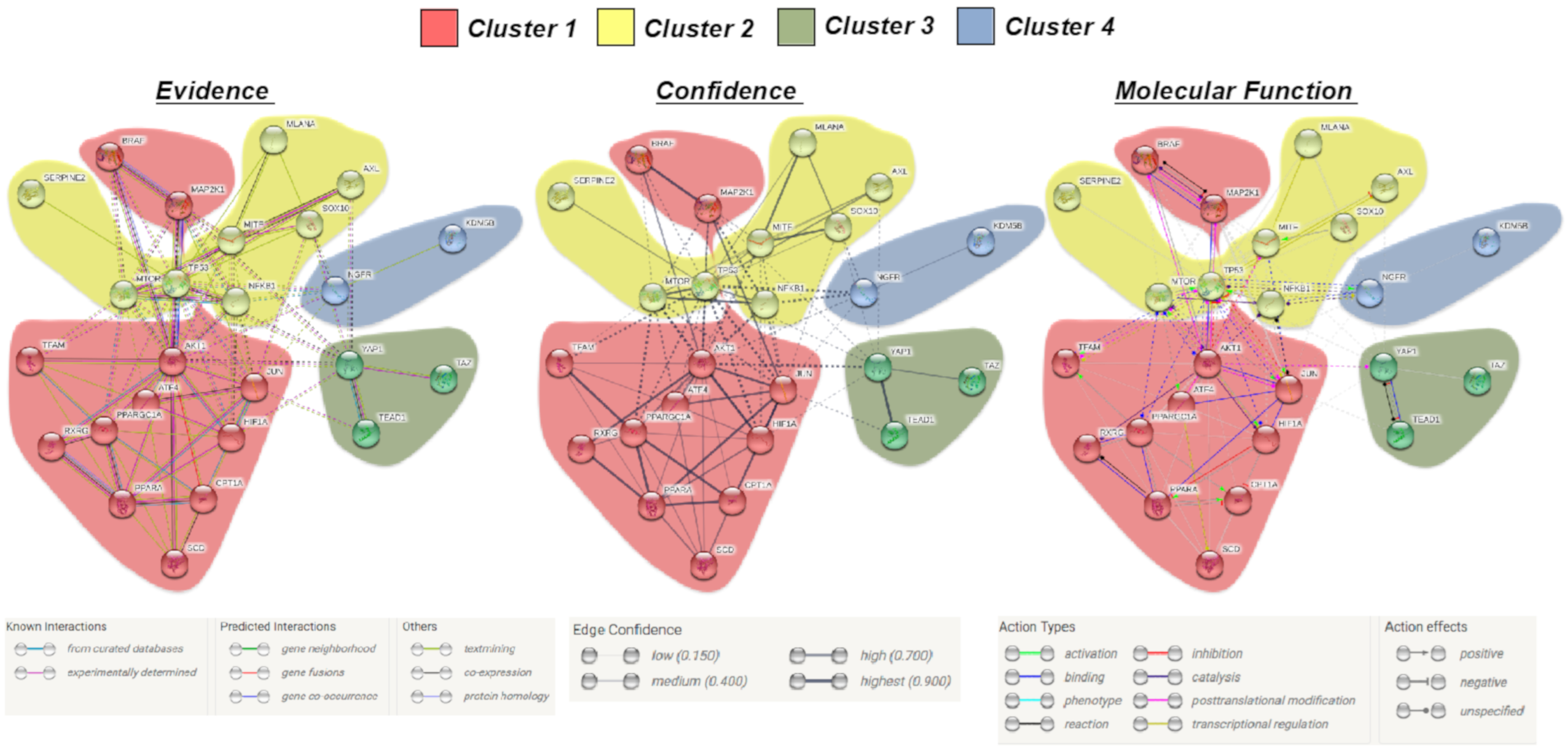
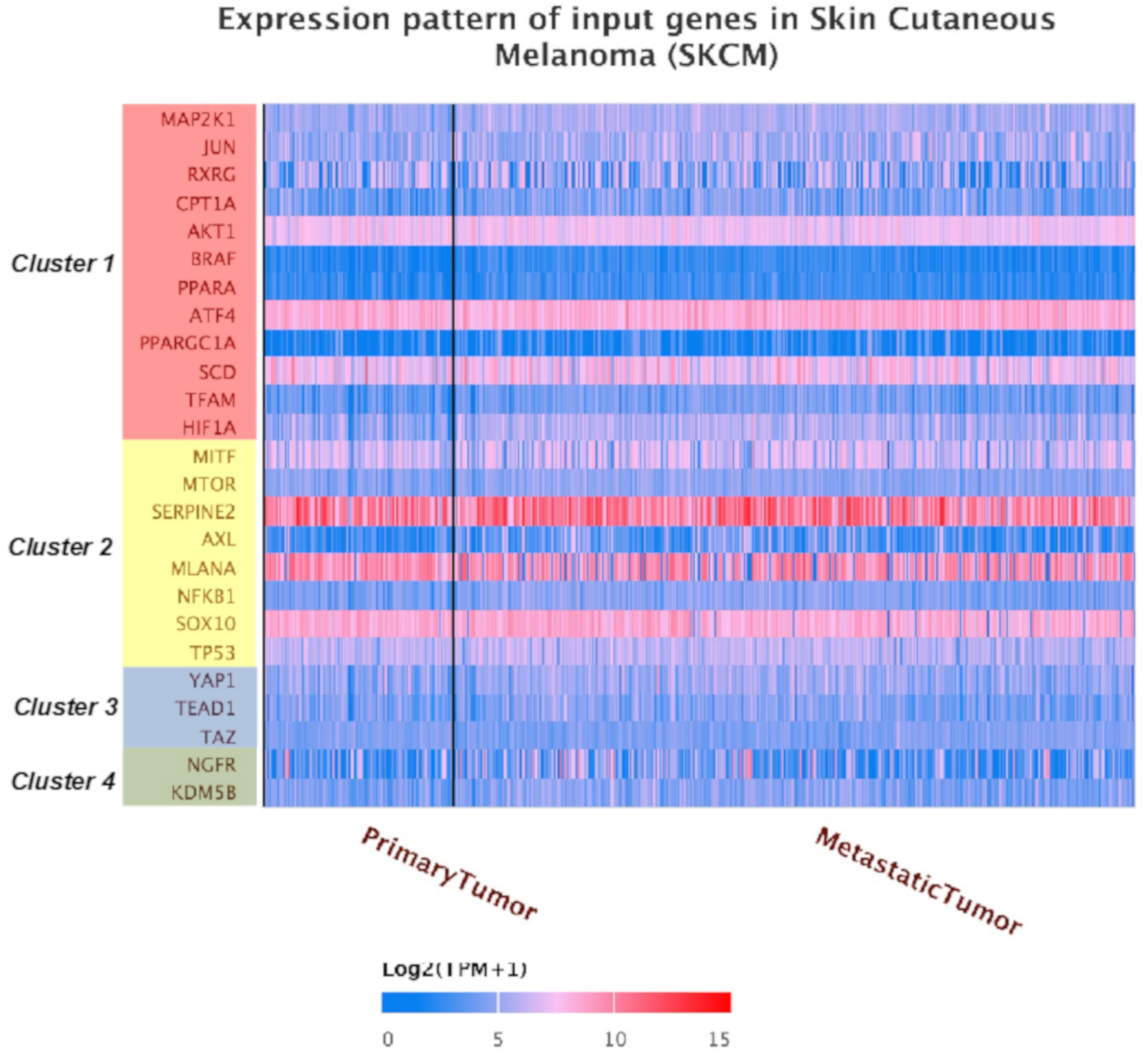
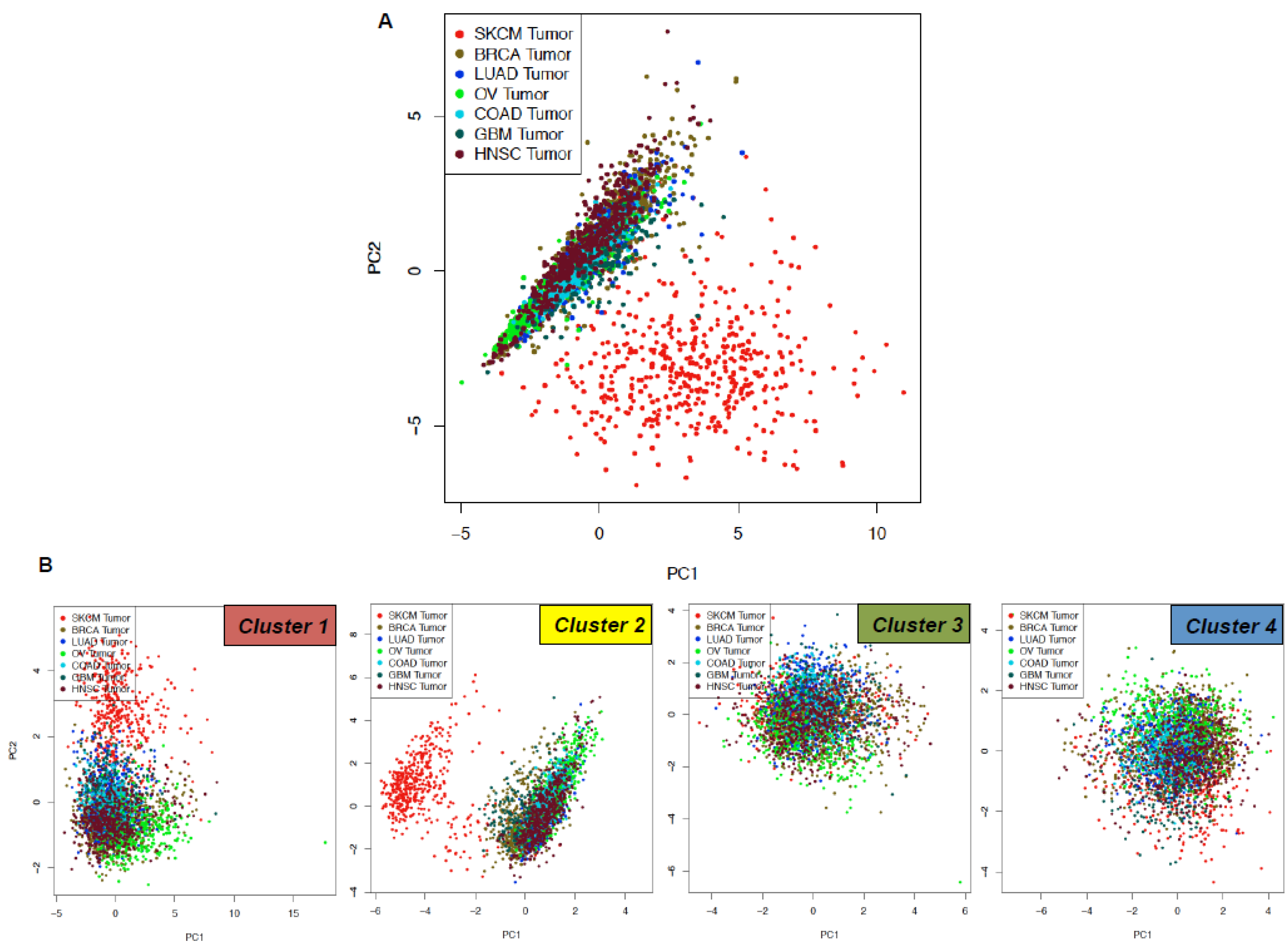
2.2. Identification of a Common Interactome Sustaining Melanoma Stem Cell Fitness Divided into Four Major Clusters
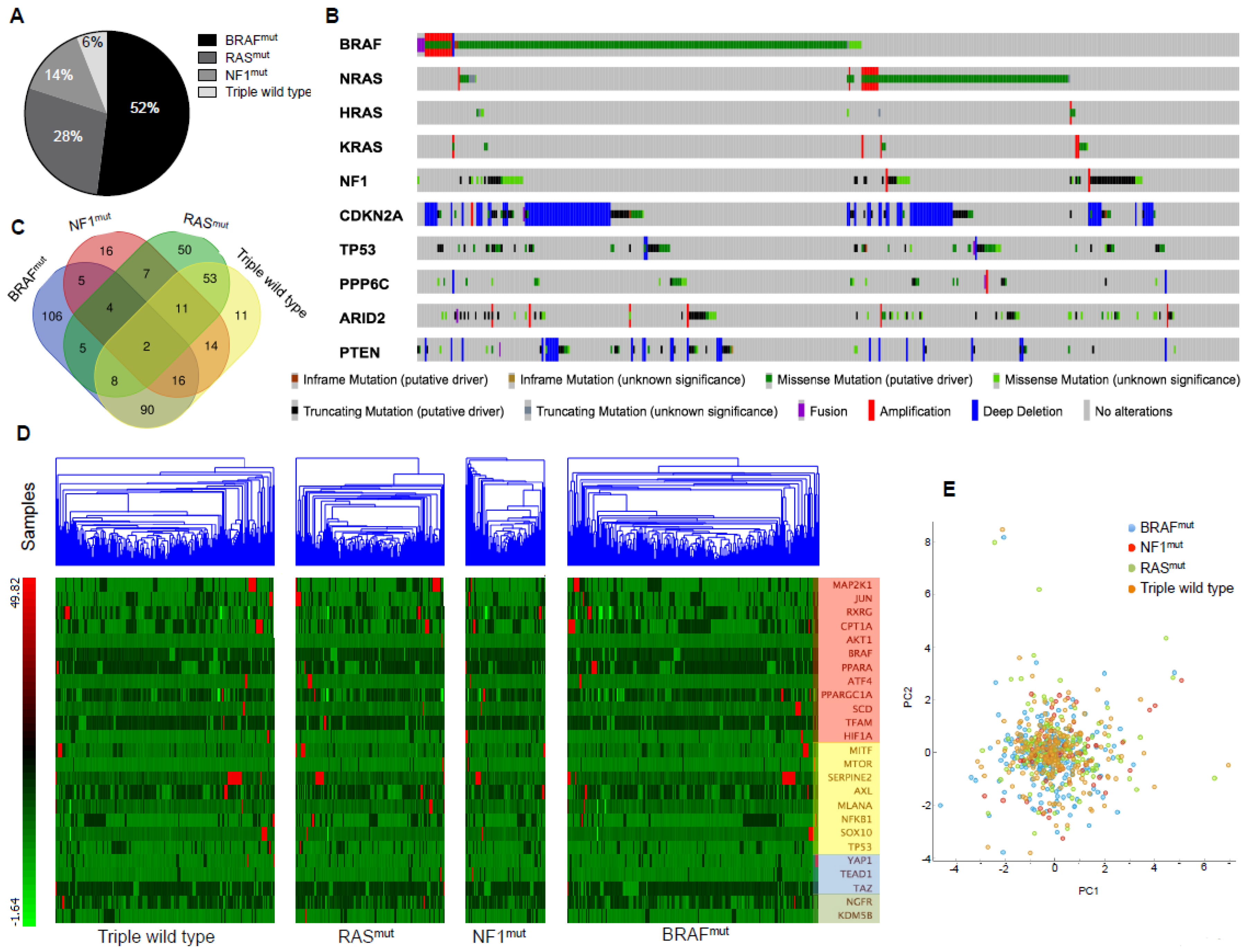
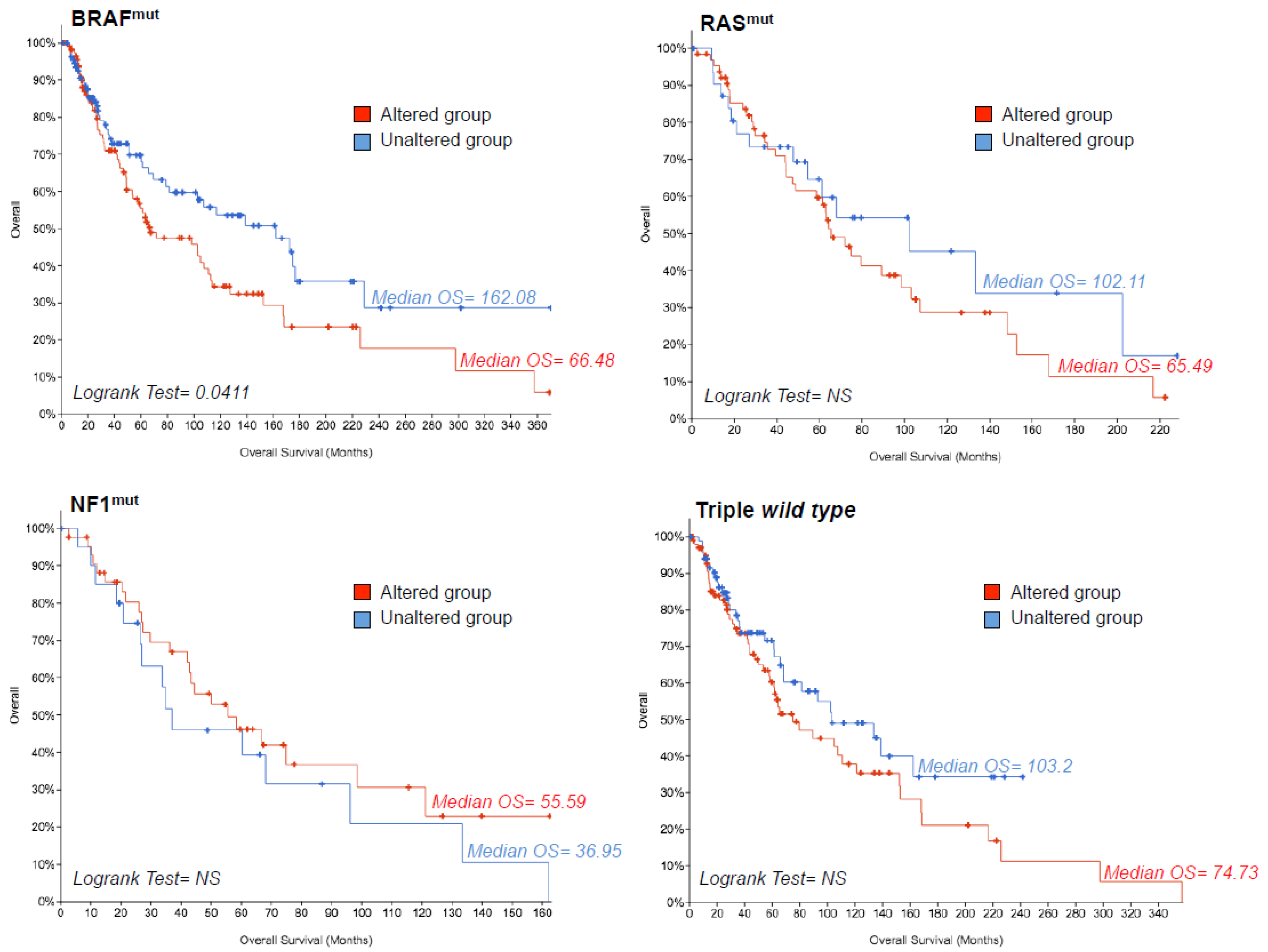
2.3. TCGA Data Mining Uncovers the Prognostic Value of “MSCsign” in BRAF-Mutant Melanomas
3. Conclusions
4. Methods
4.1. Bibliographic Search
4.2. Interactomic and Clustering Plots
4.3. Mining TCGA Data of SKCM Dataset
4.4. Principal Component Analyses
4.5. Statistical Analyses
4.6. List of the Online Tools Used
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MAPKi | BRAF/MEK inhibitors |
| CSCs | cancer stem cells |
| sc | Single Cell |
| OXPHOS | Oxidative Phosphorylation |
| MUFAs | Monounsaturated Fatty Acids |
| FAO | Fatty Acid Oxidation |
| PPI | Protein−Protein Interaction |
| MSCsign | melanoma stem cells signature |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzagalli, M.; Moretti, R.M.; Messi, E.; Marelli, M.M.; Fontana, F.; Anastasia, A.; Bani, M.R.; Beretta, G.; Limonta, P. Targeting melanoma stem cells with the Vitamin E derivative δ-tocotrienol. Sci. Rep. 2018, 8, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaviano, M.; De Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Arch. 2019, 474, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies. Eur. J. Cancer 2016, 53, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Madonna, G.; Ballesteros-Merino, C.; Feng, Z.; Bifulco, C.; Capone, M.; Giannarelli, D.; Mallardo, D.; Simeone, E.; Grimaldi, A.M.; Caracò, C.; et al. PD-L1 expression with immune-infiltrate evaluation and outcome prediction in melanoma patients treated with ipilimumab. Oncoimmunology 2018, 7, e1405206. [Google Scholar] [CrossRef] [Green Version]
- Pavlick, A.C.; Fecher, L.; Ascierto, P.A.; Sullivan, R.J. Frontline Therapy for BRAF-Mutated Metastatic Melanoma: How Do You Choose, and Is There One Correct Answer? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 564–571. [Google Scholar] [CrossRef]
- Fattore, L.; Mancini, R.; Ascierto, P.A.; Ciliberto, G. The potential of BRAF-associated non-coding RNA as a therapeutic target in melanoma. Expert Opin. Ther. Targets 2019, 23, 53–68. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G. V Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2014, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Eroglu, Z.; Infante, J.; Patel, S.; Daud, A.; Johnson, D.B.; Gonzalez, R.; Kefford, R.; Hamid, O.; Schuchter, L.; et al. Long-Term Outcomes in Patients with BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined with Trametinib. J. Clin. Oncol. 2018, 36, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting but Result in Melanoma Drug Addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, A.; Moriceau, G.; Sun, L.; Lomeli, S.; Piva, M.; Damoiseaux, R.; Holmen, S.L.; Sharpless, N.E.; Hugo, W.; Lo, R.S. Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma. Cancer Discov. 2018, 8, 74–93. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L.; Sacconi, A.; Mancini, R.; Ciliberto, G. MicroRNA-driven deregulation of cytokine expression helps development of drug resistance in metastatic melanoma. Cytokine Growth Factor Rev. 2017, 36, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.-C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [Green Version]
- Bristot, I.J.; Kehl Dias, C.; Chapola, H.; Parsons, R.B.; Klamt, F. Metabolic rewiring in melanoma drug-resistant cells. Crit. Rev. Oncol. Hematol. 2020, 153, 102995. [Google Scholar] [CrossRef]
- Ruggiero, C.F.; Malpicci, D.; Fattore, L.; Madonna, G.; Vanella, V.; Mallardo, D.; Liguoro, D.; Salvati, V.; Capone, M.; Bedogni, B.; et al. ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin. Cancers 2019, 11, 1425. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.V.; Basile, K.J.; Kugel, C.H., 3rd; Witkiewicz, A.K.; Le, K.; Amaravadi, R.K.; Karakousis, G.C.; Xu, X.; Xu, W.; Schuchter, L.M.; et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Investig. 2013, 123, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Campani, V.; Ruggiero, C.F.; Salvati, V.; Liguoro, D.; Scotti, L.; Botti, G.; Ascierto, P.A.; Mancini, R.; De Rosa, G.; et al. In Vitro Biophysical and Biological Characterization of Lipid Nanoparticles Co-Encapsulating Oncosuppressors miR-199b-5p and miR-204-5p as Potentiators of Target Therapy in Metastatic Melanoma. Int. J. Mol. Sci. 2020, 21, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattore, L.; Ruggiero, C.F.; Pisanu, M.E.; Liguoro, D.; Cerri, A.; Costantini, S.; Capone, F.; Acunzo, M.; Romano, G.; Nigita, G.; et al. Reprogramming miRNAs global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ. 2019, 26, 1267–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tupone, M.G.; D’Aguanno, S.; Di Martile, M.; Valentini, E.; Desideri, M.; Trisciuoglio, D.; Donzelli, S.; Sacconi, A.; Buglioni, S.; Ercolani, C.; et al. microRNA-378a-5p iS a novel positive regulator of melanoma progression. Oncogenesis 2020, 9, 22. [Google Scholar] [CrossRef]
- Caporali, S.; Amaro, A.; Levati, L.; Alvino, E.; Lacal, P.M.; Mastroeni, S.; Ruffini, F.; Bonmassar, L.; Antonini Cappellini, G.C.; Felli, N.; et al. miR-126-3p down-regulation contributes to dabrafenib acquired resistance in melanoma by up-regulating ADAM9 and VEGF-A. J. Exp. Clin. Cancer Res. 2019, 38, 272. [Google Scholar] [CrossRef]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Laganà, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Capone, M.; et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, E5005–E5013. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Bifulco, C.; Buonaguro, L.; Emens, L.A.; Ferris, R.L.; Fox, B.A.; Delgoffe, G.M.; Galon, J.; Gridelli, C.; Merlano, M.; et al. Perspectives in immunotherapy: Meeting report from the “Immunotherapy Bridge 2018” (28–29 November 2018, Naples, Italy). J. Immunother. Cancer 2019, 7, 332. [Google Scholar] [CrossRef]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Curran, M.A. Tumor hypermetabolism confers resistance to immunotherapy. Semin. Cancer Biol. 2020, 65, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Bruschini, S.; Ciliberto, G.; Mancini, R. The emerging role of cancer cell plasticity and cell-cycle quiescence in immune escape. Cell Death Dis. 2020, 11, 471. [Google Scholar] [CrossRef]
- De Vitis, C.; Corleone, G.; Salvati, V.; Ascenzi, F.; Pallocca, M.; De Nicola, F.; Fanciulli, M.; di Martino, S.; Bruschini, S.; Napoli, C.; et al. B4GALT1 Is a New Candidate to Maintain the Stemness of Lung Cancer Stem Cells. J. Clin. Med. 2019, 8, 1928. [Google Scholar] [CrossRef] [Green Version]
- Bruschini, S.; di Martino, S.; Pisanu, M.E.; Fattore, L.; De Vitis, C.; Laquintana, V.; Buglioni, S.; Tabbì, E.; Cerri, A.; Visca, P.; et al. CytoMatrix for a reliable and simple characterization of lung cancer stem cells from malignant pleural effusions. J. Cell. Physiol. 2020, 235, 1877–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinckerhoff, C.E. Cancer Stem Cells (CSCs) in melanoma: There’s smoke, but is there fire? J. Cell. Physiol. 2017, 232, 2674–2678. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Ahmed, F.; Haass, N.K. Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front. Oncol. 2018, 8, 173. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Ruggiero, C.F.; Liguoro, D.; Mancini, R.; Ciliberto, G. Single cell analysis to dissect molecular heterogeneity and disease evolution in metastatic melanoma. Cell Death Dis. 2019, 10, 827. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Fisher, D.E.; Flaherty, K.T. Cell-state dynamics and therapeutic resistance in melanoma from the perspective of MITF and IFNγ pathways. Nat. Rev. Clin. Oncol. 2019, 16, 549–562. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Sini, M.C.; Doneddu, V.; Paliogiannis, P.; Casula, M.; Colombino, M.; Manca, A.; Botti, G.; Ascierto, P.A.; Lissia, A.; Cossu, A.; et al. Genetic alterations in main candidate genes during melanoma progression. Oncotarget 2018, 9, 8531–8541. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [Green Version]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Papucci, L.; Magnelli, L.; Mazzanti, B.; Stecca, B.; Calorini, L. The acidic tumor microenvironment drives a stem-like phenotype in melanoma cells. J. Mol. Med. 2020, 98, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Gerber, T.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Schadendorf, D.; Schartl, M.; Anderegg, U.; Camp, G.; Treutlein, B.; Binder, H.; et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget 2017, 8, 846–862. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Aloia, A.; Müllhaupt, D.; Chabbert, C.D.; Eberhart, T.; Flückiger-Mangual, S.; Vukolic, A.; Eichhoff, O.; Irmisch, A.; Alexander, L.T.; Scibona, E.; et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors. Clin. Cancer Res. 2019, 25, 6852–6867. [Google Scholar] [CrossRef]
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 2020, 77, 120–137.e9. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, Y.N.V.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, F.C.E.; Bordag, N.; Zügner, E.; Trajkovic-Arsic, M.; Chauvistré, H.; Shannan, B.; Váraljai, R.; Horn, S.; Magnes, C.; Thomas Siveke, J.; et al. Targeting the H3K4 Demethylase KDM5B Reprograms the Metabolome and Phenotype of Melanoma Cells. J. Investig. Dermatol. 2019, 139, 2506–2516.e10. [Google Scholar] [CrossRef] [PubMed]
- Pisanu, M.E.; Maugeri-Saccà, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbì, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 318. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ko, M.E.; Cheng, H.; Zhu, R.; Xue, M.; Wang, J.; Lee, J.W.; Frankiw, L.; Xu, A.; Wong, S.; et al. Multi-omic single-cell snapshots reveal multiple independent trajectories to drug tolerance in a melanoma cell line. Nat. Commun. 2020, 11, 2345. [Google Scholar] [CrossRef]
- Vashisht Gopal, Y.N.; Gammon, S.; Prasad, R.; Knighton, B.; Pisaneschi, F.; Roszik, J.; Feng, N.; Johnson, S.; Pramanik, S.; Sudderth, J.; et al. A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin. Cancer Res. 2019, 25, 6429–6442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perego, M.; Maurer, M.; Wang, J.X.; Shaffer, S.; Müller, A.C.; Parapatics, K.; Li, L.; Hristova, D.; Shin, S.; Keeney, F.; et al. A slow-cycling subpopulation of melanoma cells with highly invasive properties. Oncogene 2018, 37, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical Role for Wild-Type p53 in Driving Therapy Resistance in Melanoma. Mol. Cell 2020, 77, 633–644.e5. [Google Scholar] [CrossRef]
- Zubrilov, I.; Sagi-Assif, O.; Izraely, S.; Meshel, T.; Ben-Menahem, S.; Ginat, R.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.-O.; Hoon, D.S.B.; et al. Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett. 2015, 361, 86–96. [Google Scholar] [CrossRef]
- Liguoro, D.; Fattore, L.; Mancini, R.; Ciliberto, G. Drug tolerance to target therapy in melanoma revealed at single cell level: What next? Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188440. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Zakaria, N.; Mohd Yusoff, N.; Zakaria, Z.; Widera, D.; Yahaya, B.H. Inhibition of NF-κB Signaling Reduces the Stemness Characteristics of Lung Cancer Stem Cells. Front. Oncol. 2018, 8, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, H.; Moriyama, M.; Ozawa, T.; Tsuruta, D.; Iguchi, T.; Tamada, S.; Nakatani, T.; Nakagawa, K.; Hayakawa, T. Notch Signaling Enhances Stemness by Regulating Metabolic Pathways Through Modifying p53, NF-κB, and HIF-1α. Stem Cells Dev. 2018, 27, 935–947. [Google Scholar] [CrossRef]
- Fattore, L.; Malpicci, D.; Milite, C.; Castellano, S.; Sbardella, G.; Botti, G.; Ascierto, P.A.; Mancini, R.; Ciliberto, G. Reverse transcriptase inhibition potentiates target therapy in BRAF-mutant melanomas: Effects on cell proliferation, apoptosis, DNA-damage, ROS induction and mitochondrial membrane depolarization. Cell Commun. Signal. 2020, 18, 150. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Giarnieri, E.; De Vitis, C.; Malanga, D.; Roscilli, G.; Noto, A.; Marra, E.; Laudanna, C.; Zoppoli, P.; De Luca, P.; et al. Spheres derived from lung adenocarcinoma pleural effusions: Molecular characterization and tumor engraftment. PLoS ONE 2011, 6, e21320. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; Raffa, S.; De Vitis, C.; Roscilli, G.; Malpicci, D.; Coluccia, P.; Di Napoli, A.; Ricci, A.; Giovagnoli, M.R.; Aurisicchio, L.; et al. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013, 4, e947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Saccà, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; De Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef]
- Knobloch, M.; Pilz, G.-A.; Ghesquière, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Alicea, G.M.; Rebecca, V.W.; Goldman, A.R.; Fane, M.E.; Douglass, S.M.; Behera, R.; Webster, M.R.; Kugel, C.H., 3rd; Ecker, B.L.; Caino, M.C.; et al. Changes in Aged Fibroblast Lipid Metabolism Induce Age-Dependent Melanoma Cell Resistance to Targeted Therapy via the Fatty Acid Transporter FATP2. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; et al. Cancer-selective targeting of the NF-κB survival pathway with GADD45β/MKK7 inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Pippa, S.; Mannironi, C.; Licursi, V.; Bombardi, L.; Colotti, G.; Cundari, E.; Mollica, A.; Coluccia, A.; Naccarato, V.; La Regina, G.; et al. Small Molecule Inhibitors of KDM5 Histone Demethylases Increase the Radiosensitivity of Breast Cancer Cells Overexpressing JARID1B. Molecules 2019, 24, 1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description | Reference # | Cluster |
|---|---|---|---|
| MAP2K1 | Dual specificity mitogen-activated protein kinase kinase 1 | [9,11,12,13,14,15] | Kinase and metabolic signature |
| JUN | Transcription factor AP-1 | [15,42,46,56,58] | |
| RXRG | Retinoic acid receptor RXR-gamma | [42,53] | |
| CPT1A | Carnitine O-palmitoyltransferase 1 | [59] | |
| AKT1 | RAC-alpha serine/threonine-protein kinase | [9,16,20] | |
| BRAF | Serine/threonine-protein kinase B-raf | [4,7,9,11,12,13,14] | |
| PPARA | Peroxisome proliferator-activated receptor alpha | [59] | |
| ATF4 | Cyclic AMP-dependent transcription factor ATF-4 | [20,60] | |
| PPARGC1A | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha | [20,61,62,63] | |
| SCD | Acyl-CoA desaturase | [60,64] | |
| TFAM | Transcription factor A, mitochondrial | [65] | |
| HIF1A | Hypoxia-inducible factor 1-alpha | [20,66] | |
| MITF | Microphthalmia-associated transcription factor | [20,42,46,47,50,52,53,56,57,60,61,62,64,67] | Melanoma-associated signature |
| MTOR | Serine/threonine-protein kinase mTOR | [62,67] | |
| SERPINE2 | Glia-derived nexin | [68] | |
| AXL | Tyrosine-protein kinase receptor UFO | [20,42,46,47,50,52,53,56,57,58,60,66] | |
| MLANA | Melanoma antigen recognized by T-cells 1 | [52,53,56,66] | |
| NFKB1 | Nuclear factor NF-kappa-B p105 subunit | [52,66] | |
| SOX10 | Transcription factor SOX-10 | [53,58] | |
| TP53 | Cellular tumor antigen p53 | [69] | |
| YAP1 | Transcriptional coactivator YAP1 | [47,64] | Hippo pathway signature |
| TEAD1 | Transcriptional enhancer factor TEF-1 | [47,58] | |
| TAZ | Tafazzin | [47,64] | |
| NGFR | Tumor necrosis factor receptor superfamily member 16 | [52,53,54,56,57,58,66,70,71] | Slow cycling signature |
| KDM5B | Lysine-specific demethylase 5B | [55,56,63,64,66,70,72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fattore, L.; Mancini, R.; Ciliberto, G. Cancer Stem Cells and the Slow Cycling Phenotype: How to Cut the Gordian Knot Driving Resistance to Therapy in Melanoma. Cancers 2020, 12, 3368. https://doi.org/10.3390/cancers12113368
Fattore L, Mancini R, Ciliberto G. Cancer Stem Cells and the Slow Cycling Phenotype: How to Cut the Gordian Knot Driving Resistance to Therapy in Melanoma. Cancers. 2020; 12(11):3368. https://doi.org/10.3390/cancers12113368
Chicago/Turabian StyleFattore, Luigi, Rita Mancini, and Gennaro Ciliberto. 2020. "Cancer Stem Cells and the Slow Cycling Phenotype: How to Cut the Gordian Knot Driving Resistance to Therapy in Melanoma" Cancers 12, no. 11: 3368. https://doi.org/10.3390/cancers12113368
APA StyleFattore, L., Mancini, R., & Ciliberto, G. (2020). Cancer Stem Cells and the Slow Cycling Phenotype: How to Cut the Gordian Knot Driving Resistance to Therapy in Melanoma. Cancers, 12(11), 3368. https://doi.org/10.3390/cancers12113368