Immunoprofiling in Neuroendocrine Neoplasms Unveil Immunosuppressive Microenvironment

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
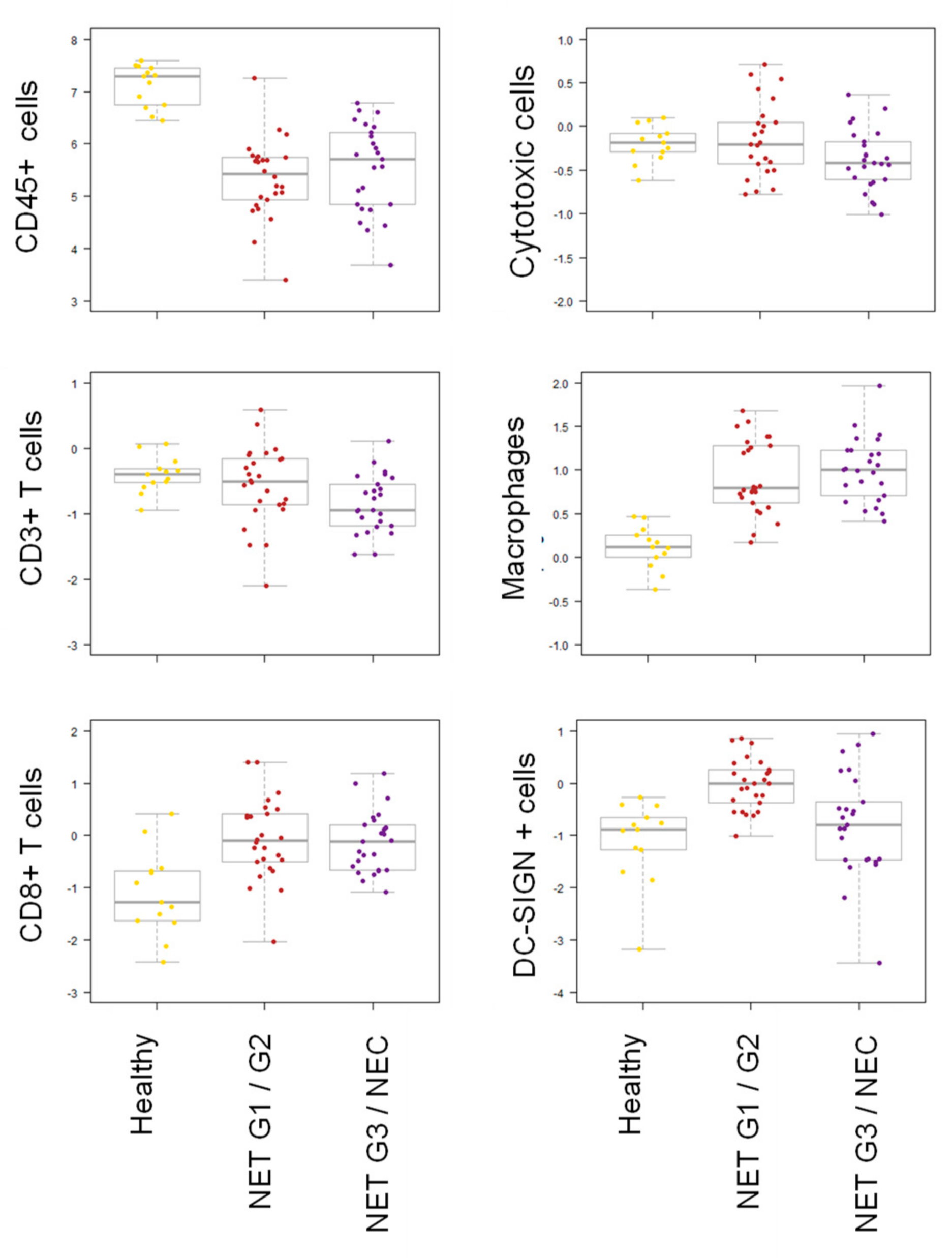
2.2. Lower Intratumoral CD3+ T Cell Infiltration in NET G1/G2 Compared to NETG3/NEC
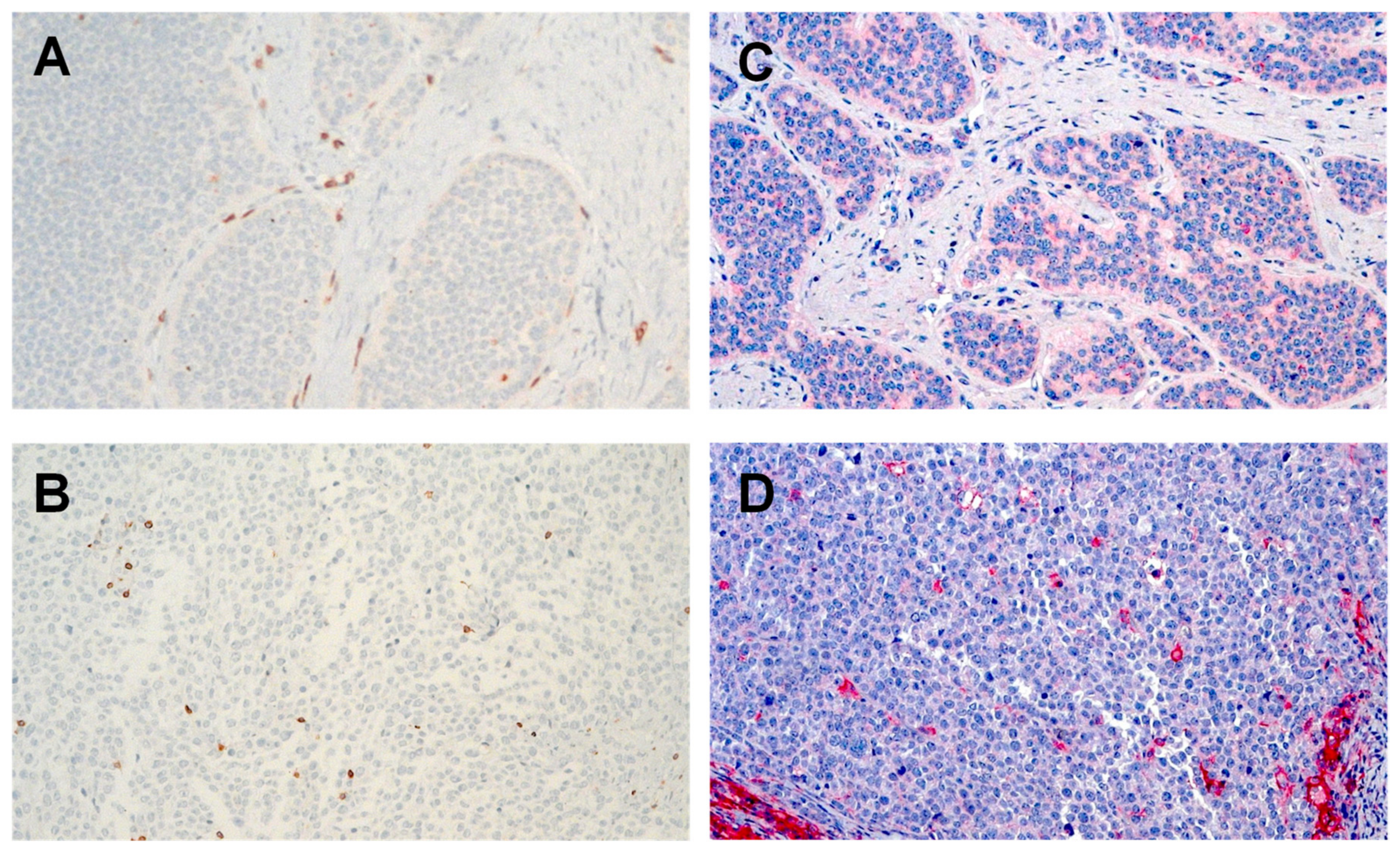
2.3. PD-L1 Is Expressed Higher in NET G3/NEC than NET G1/G2
2.4. Low PD-1 and TIM-3 Expression on Stromal and Intratumoral T Cells
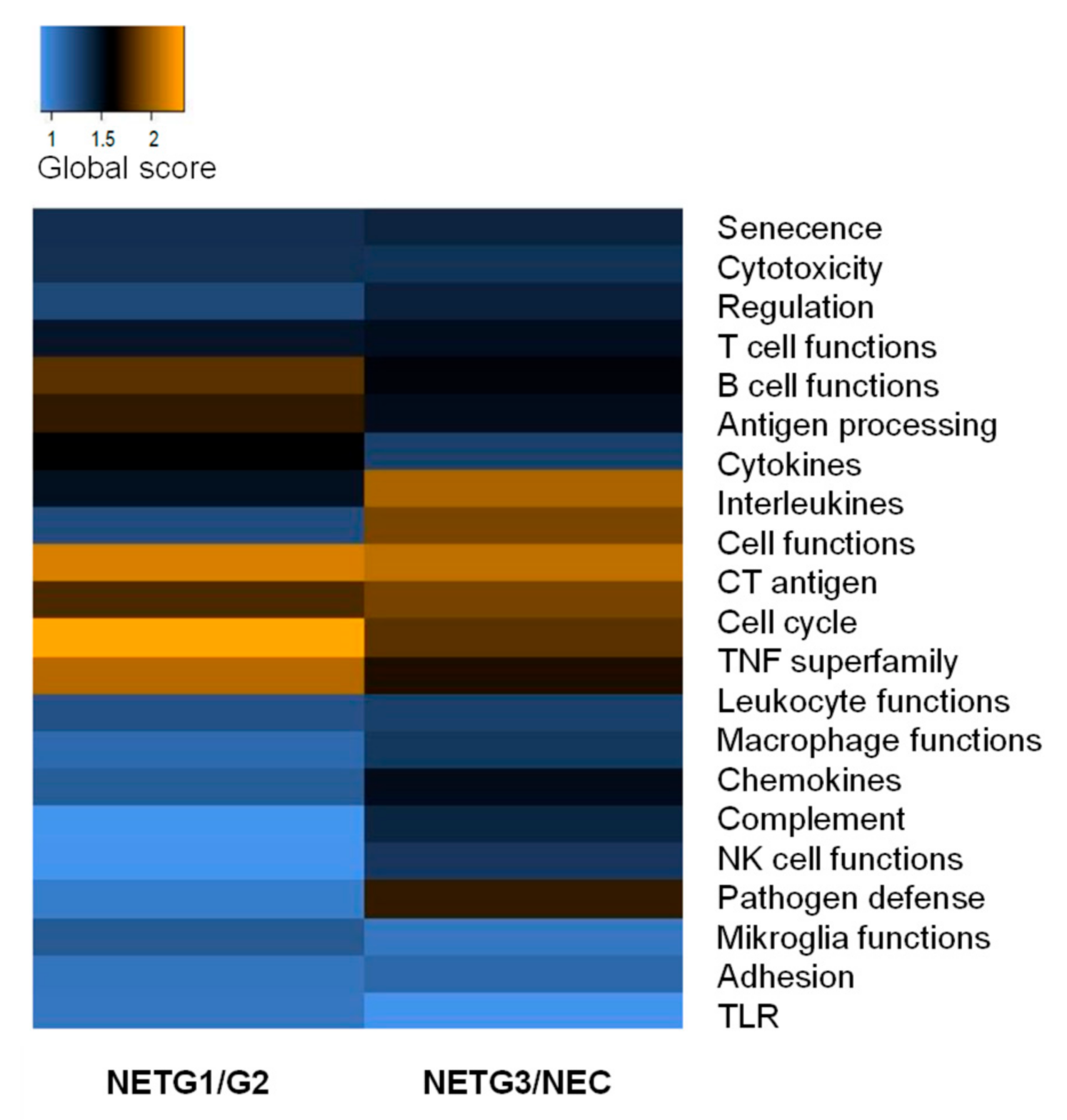
2.5. mRNA Immunoprofiling Showed Differential Immune Response in NET G1/G2 and NET G3/NEC
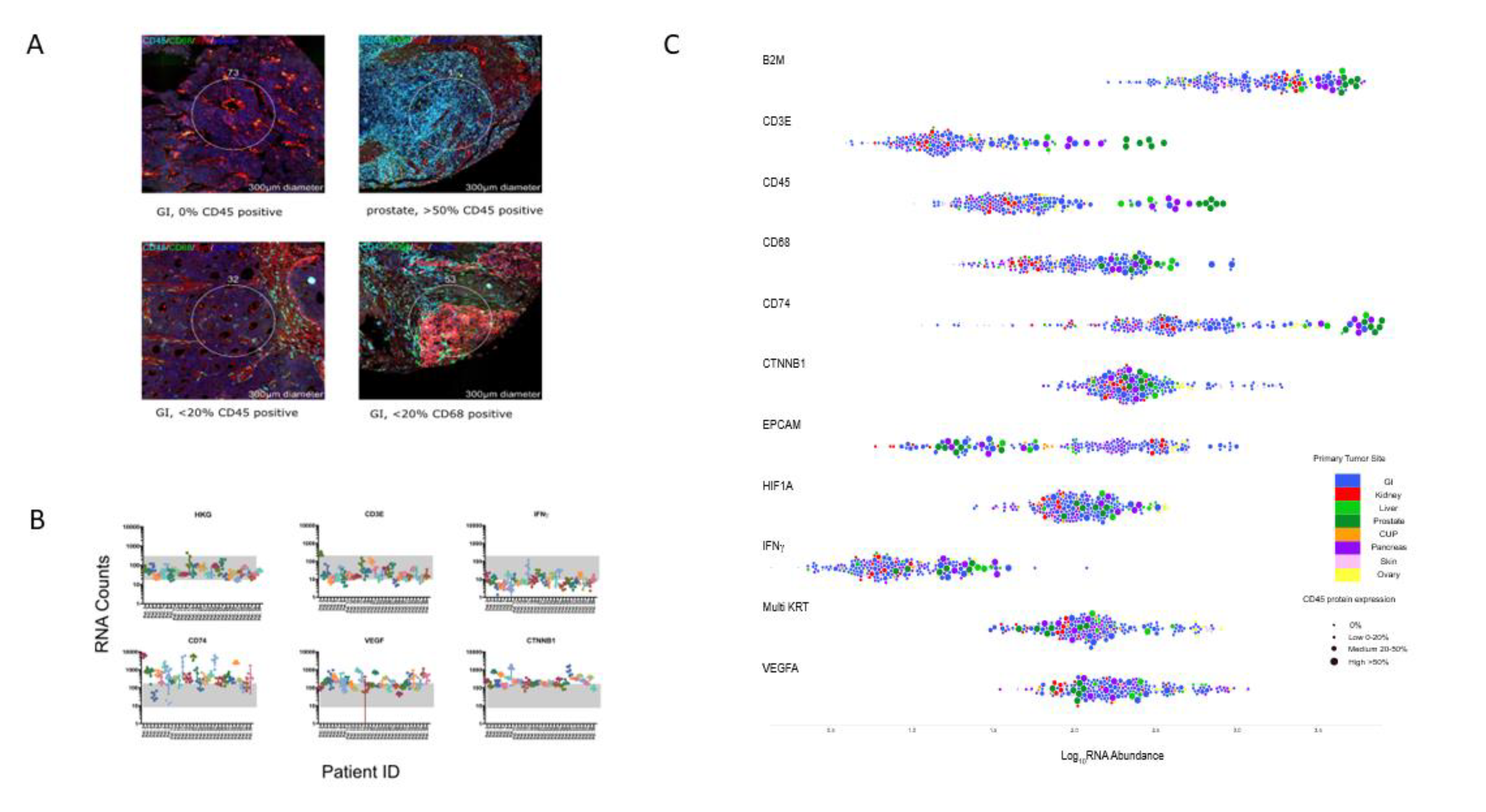
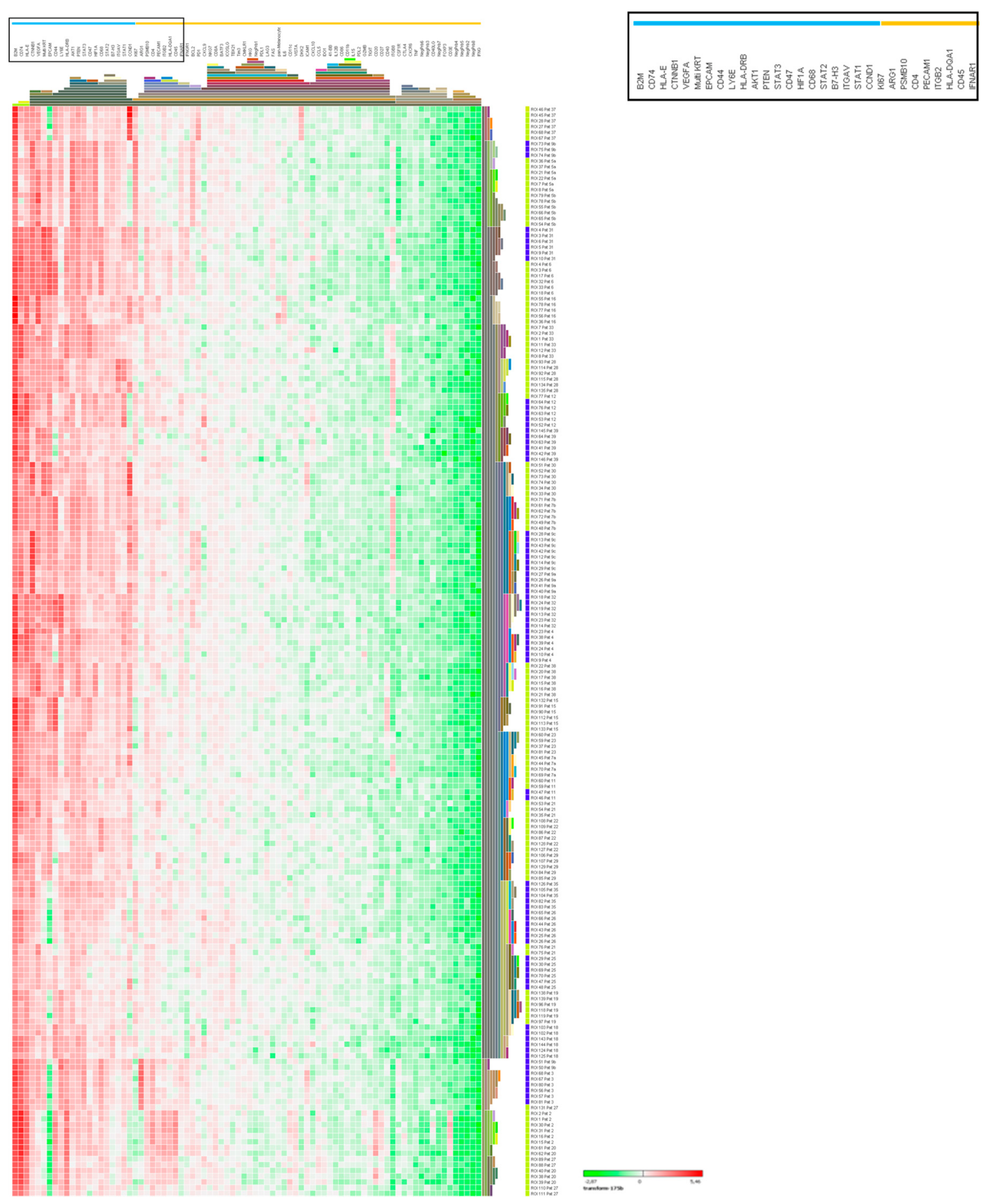
2.6. Digital Spatial Profiling of NEC and NET G3 Samples
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Immunohistochemistry
4.3. Extraction of RNA
4.4. mRNA Immunoprofiling (NanoString)
4.5. Digital Spatial Profiling
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.C. Tim-3: An emerging target in the cancer immunotherapy landscape. Cancer Immunol. Res. 2014, 2, 393–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Horsch, D.; Grabowski, P.; Schneider, C.P.; Petrovitch, A.; Kaemmerer, D.; Hommann, M.; Baum, R.P. Current treatment options for neuroendocrine tumors. Drugs Today 2011, 47, 773–786. [Google Scholar] [CrossRef]
- Auernhammer, C.J.; Spitzweg, C.; Angele, M.K.; Boeck, S.; Grossman, A.; Nolting, S.; Ilhan, H.; Knosel, T.; Mayerle, J.; Reincke, M.; et al. Advanced neuroendocrine tumours of the small intestine and pancreas: Clinical developments, controversies, and future strategies. Lancet Diabetes Endocrinol. 2018, 6, 404–415. [Google Scholar] [CrossRef]
- Yao, J.C.; Strosberg, J.; Fazio, N.; Pavel, M.; Ruszniewski, P.; Bergsland, E.; Li, D.; Tafuto, S.; Raj, N.; Campana, D.; et al. Activity & Safety of Spartalizumab (PDR001) in Patients (pts) With Advanced Neuroendocrine Tumors (NET) of Pancreatic (Pan), Gastrointestinal (GI), or Thoracic (T) Origin, & Gastroenteropancreatic Neuroendocrine Carcinoma (GEP NEC) Who Have Progressed on Prior Treatment (Tx). Ann. Oncol. 2018, 29 (Suppl. S8), viii467–viii478. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.-P.; Shapira-Frommer, R.; Bergsland, E.K.; Shah, M.H.; Fakih, M.; Takahashi, S.; et al. Pembrolizumab treatment of advanced neuroendocrine tumors: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2019, 37, 190. [Google Scholar] [CrossRef]
- Vijayvergia, N.; Dasari, A.; Ross, E.A.; Dotan, E.; Halperin, D.M.; Astsaturov, I.A.; Hall, M.J.; Ross, N.M.; McClean, D.; Denlinger, C.S.; et al. Pembrolizumab (P) monotherapy in patients with previously treated metastatic high grade neuroendocrine neoplasms (HG-NENs). J. Clin. Oncol. 2018, 36, 4104. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Othus, M.; Chae, Y.K.; Giles, F.J.; Hansel, D.E.; Singh, P.P.; Fontaine, A.; Shah, M.H.; Kasi, A.; Baghdadi, T.A.; et al. A Phase II Basket Trial of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART SWOG 1609) in Patients with Nonpancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2020, 26, 2290–2296. [Google Scholar] [CrossRef] [Green Version]
- Capdevila, J.; Teule, A.; López, C.; García-Carbonero, R.; Benavent, M.; Custodio, A.; Cubillo, A.; Alonso, V.; Alonso Gordoa, T.; Carmona-Bayonas, A.; et al. A multi-cohort phase II study of durvalumab plus tremelimumab for the treatment of patients (pts) with advanced neuroendocrine neoplasms (NENs) of gastroenteropancreatic or lung origin: The DUNE trial (GETNE 1601). Ann. Oncol. 2020, 31 (Suppl. S4), S711–S724. [Google Scholar] [CrossRef]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Danilova, L.; Wang, H.; Sunshine, J.; Kaunitz, G.J.; Cottrell, T.R.; Xu, H.; Esandrio, J.; Anders, R.A.; Cope, L.; Pardoll, D.M.; et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc. Natl. Acad. Sci. USA 2016, 113, E7769–E7777. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F. HLA Class-I Expression and Cancer Immunotherapy. Adv. Exp. Med. Biol. 2019, 1151, 79–90. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Meng, Y.; Blank, C.; Brown, I.; Kacha, A.; Kline, J.; Harlin, H. Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev. 2006, 213, 131–145. [Google Scholar] [CrossRef]
- Schouppe, E.; De Baetselier, P.; Van Ginderachter, J.A.; Sarukhan, A. Instruction of myeloid cells by the tumor microenvironment: Open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology 2012, 1, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.C.; Donkor, C.; Glasgow, K.; Pillarisetty, V.G.; Gonen, M.; Espat, N.J.; Klimstra, D.S.; D’Angelica, M.I.; Allen, P.J.; Jarnagin, W.; et al. T cell infiltrate and outcome following resection of intermediate-grade primary neuroendocrine tumours and liver metastases. HPB 2010, 12, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikman, S.; Sommaggio, R.; De La Torre, M.; Oberg, K.; Essand, M.; Giandomenico, V.; Loskog, A.; Totterman, T.H. Midgut carcinoid patients display increased numbers of regulatory T cells in peripheral blood with infiltration into tumor tissue. Acta Oncol. 2009, 48, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryschich, E.; Autschbach, F.; Eisold, S.; Klar, E.; Buchler, M.W.; Schmidt, J. Expression of HLA class I/II antigens and T cell immune response in human neuroendocrine tumors of the pancreas. Tissue Antigens 2003, 62, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Ferrata, M.; Schad, A.; Zimmer, S.; Musholt, T.J.; Bahr, K.; Kuenzel, J.; Becker, S.; Springer, E.; Roth, W.; Weber, M.M.; et al. PD-L1 Expression and Immune Cell Infiltration in Gastroenteropancreatic (GEP) and Non-GEP Neuroendocrine Neoplasms With High Proliferative Activity. Front. Oncol. 2019, 9, 343. [Google Scholar] [CrossRef] [Green Version]
- Krug, S.; Abbassi, R.; Griesmann, H.; Sipos, B.; Wiese, D.; Rexin, P.; Blank, A.; Perren, A.; Haybaeck, J.; Huttelmaier, S.; et al. Therapeutic targeting of tumor-associated macrophages in pancreatic neuroendocrine tumors. Int. J. Cancer 2018, 143, 1806–1816. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Michelakos, T.; Deshpande, V.; Arora, K.S.; Yamada, T.; Ting, D.T.; Taylor, M.S.; Castillo, C.F.; Warshaw, A.L.; Lillemoe, K.D.; et al. Role of Tumor-Associated Macrophages in the Clinical Course of Pancreatic Neuroendocrine Tumors (PanNETs). Clin. Cancer Res. 2019, 25, 2644–2655. [Google Scholar] [CrossRef]
- Cavalcanti, E.; Armentano, R.; Valentini, A.M.; Chieppa, M.; Caruso, M.L. Role of PD-L1 expression as a biomarker for GEP neuroendocrine neoplasm grading. Cell Death Dis. 2017, 8, e3004. [Google Scholar] [CrossRef]
- Kim, S.T.; Ha, S.Y.; Lee, S.; Ahn, S.; Lee, J.; Park, S.H.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Kim, K.M.; et al. The Impact of PD-L1 Expression in Patients with Metastatic GEP-NETs. J. Cancer 2016, 7, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Bethmann, D.; Feng, Z.; Fox, B.A. Immunoprofiling as a predictor of patient’s response to cancer therapy-promises and challenges. Curr. Opin. Immunol. 2017, 45, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Bruwer, K.; Altendorf-Hofmann, A.; Auernhammer, C.J.; Spitzweg, C.; Westphalen, C.B.; Boeck, S.; Schubert-Fritschle, G.; Werner, J.; Heinemann, V.; et al. Immune checkpoint markers in gastroenteropancreatic neuroendocrine neoplasia. Endocr.-Relat. Cancer 2019, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Strosberg, J.; Al Diffalha, S.; Coppola, D. Analysis of the immune landscape of small bowel neuroendocrine tumors. Endocr.-Relat. Cancer 2019, 26, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Sampedro-Nunez, M.; Serrano-Somavilla, A.; Adrados, M.; Cameselle-Teijeiro, J.M.; Blanco-Carrera, C.; Cabezas-Agricola, J.M.; Martinez-Hernandez, R.; Martin-Perez, E.; Munoz de Nova, J.L.; Diaz, J.A.; et al. Analysis of expression of the PD-1/PD-L1 immune checkpoint system and its prognostic impact in gastroenteropancreatic neuroendocrine tumors. Sci. Rep. 2018, 8, 17812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, A.; Bowden, M.; Zhang, S.; Masugi, Y.; Thorner, A.R.; Herbert, Z.T.; Zhou, C.W.; Brais, L.; Chan, J.A.; Hodi, F.S.; et al. Characterization of the Neuroendocrine Tumor Immune Microenvironment. Pancreas 2018, 47, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Atefi, M.; Avramis, E.; Lassen, A.; Wong, D.J.; Robert, L.; Foulad, D.; Cerniglia, M.; Titz, B.; Chodon, T.; Graeber, T.G.; et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin. Cancer Res. 2014, 20, 3446–3457. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014, 25, 590–604. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Vetizou, M.; Daillere, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, D.; Kojima, M.; Suzuki, T.; Sugimoto, M.; Kobayashi, S.; Takahashi, S.; Konishi, M.; Gotohda, N.; Ikeda, M.; Nakatsura, T.; et al. Profiling the Tumour Immune Microenvironment in Pancreatic Neuroendocrine Neoplasms with Multispectral Imaging Indicates Distinct Subpopulation Characteristics Concordant with WHO 2017 Classification. Sci. Rep. 2018, 8, 13166. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Mafficini, A.; Scarpa, A. Genetics and Epigenetics of Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr. Rev. 2019, 40, 506–536. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Murray, P.J. Understanding local macrophage phenotypes in disease: Modulating macrophage function to treat cancer. Nat. Med. 2015, 21, 117–119. [Google Scholar] [CrossRef]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Geis, C.; Fendrich, V.; Rexin, P.; Di Fazio, P.; Bartsch, D.K.; Ocker, M.; Quint, K.; Heverhagen, A.E. Ileal neuroendocrine tumors show elevated activation of mammalian target of rapamycin complex. J. Surg. Res. 2015, 194, 388–393. [Google Scholar] [CrossRef]
- Banck, M.S.; Kanwar, R.; Kulkarni, A.A.; Boora, G.K.; Metge, F.; Kipp, B.R.; Zhang, L.; Thorland, E.C.; Minn, K.T.; Tentu, R.; et al. The genomic landscape of small intestine neuroendocrine tumors. J. Clin. Investig. 2013, 123, 2502–2508. [Google Scholar] [CrossRef] [Green Version]
- Briest, F.; Grabowski, P. PI3K-AKT-mTOR-signaling and beyond: The complex network in gastroenteropancreatic neuroendocrine neoplasms. Theranostics 2014, 4, 336–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottarelli, L.; Azzoni, C.; Pizzi, S.; D’Adda, T.; Silini, E.M.; Bordi, C.; Rindi, G. Adenomatous polyposis coli gene involvement in ileal enterochromaffin cell neuroendocrine neoplasms. Hum. Pathol. 2013, 44, 2736–2742. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/beta-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis 2013, 34, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briest, F.; Berg, E.; Grass, I.; Freitag, H.; Kaemmerer, D.; Lewens, F.; Christen, F.; Arsenic, R.; Altendorf-Hofmann, A.; Kunze, A.; et al. FOXM1: A novel drug target in gastroenteropancreatic neuroendocrine tumors. Oncotarget 2015, 6, 8185–8199. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 2016, 5, e1086862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Ihara, S.; Kida, H.; Arase, H.; Tripathi, L.P.; Chen, Y.A.; Kimura, T.; Yoshida, M.; Kashiwa, Y.; Hirata, H.; Fukamizu, R.; et al. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis. Cancer Res. 2012, 72, 2990–2999. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [Green Version]
- WHO. Classification of Tumours, 4th ed.; WHO: Geneva, Switzerland, 2017; Volume 10. [Google Scholar]
- Therasse, P. Measuring the clinical response. What does it mean? Eur. J. Cancer 2002, 38, 1817–1823. [Google Scholar] [CrossRef]
- Fortina, P.; Surrey, S. Digital mRNA profiling. Nat. Biotechnol. 2008, 26, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, B.; Wang, M. CD47 is a novel potent immunotherapy target in human malignancies: Current studies and future promises. Future Oncol. 2018, 14, 2179–2188. [Google Scholar] [CrossRef]
- Figueiredo, C.R.; Azevedo, R.A.; Mousdell, S.; Resende-Lara, P.T.; Ireland, L.; Santos, A.; Girola, N.; Cunha, R.; Schmid, M.C.; Polonelli, L.; et al. Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response Against Metastatic Melanoma. Front. Immunol. 2018, 9, 1132. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | |
|---|---|
| Patient samples available | 78 |
| Sex available | 73 |
| Male/female | 37/36 |
| Median age | 64 (30–84) |
| WHO grading classification available | 78 |
| NET (G1/G2) | 48 (36/12) |
| NEC/NET G3 | 30 (20/10) |
| Stage at time of tissue collection | |
| NET (G1/G2) | |
| II/III/IV | 1/9/38 |
| NEC/NET G3 | |
| II/III/IV | 6/7/17 |
| Primary tumor localization (n) | |
| NET (G1/G2) | Ileum (47), pancreas (1) |
| NEC/NET G3 | Gastrointestinal (17), bile duct (3), gall bladder (1), pancreas (5), bladder (1), kidney (1), CUP (2) |
| Tissue analyzed Primary tumor/metastasis | |
| NET (G1/G2) | 48/0 |
| NEC/NET G3 | 24/6 |
| Localization of analyzed metastasis | Lymph node (3), liver (2), skin (1) |
| NET G1/G2 | NET G3/NEC | |
|---|---|---|
| CD3+ T cell infiltration | ||
| Patient samples available | 41 | 29 |
| Stromal T cell infiltration Median counts/10 HPF | 60 (range 25–87) | 58 (range 18–150) |
| Intratumoral T cell infiltration Median counts/10 HPF | 9/10 HPF (range 4–51) | 16/10 HPF (range 3–75) |
| TIM-3 expression | ||
| Stromal lymphocytes Positive samples | 20/36 (56%) | 16/23 (70%) |
| Intratumoral lymphocytes Stained positive | 1/36 (3%) | 5/23 (22%) |
| PD-1/PD-L1 expression | ||
| PD-L1-Expression on tumor cells Expression level >1% on cell membrane | 11/46 (24%) | 15/30 (50%) |
| PD-1-Expression on lymphocytes Expression level >1% on cell membrane | 0/42 (0%) | 2/17 (12%) |
| Genes | Log2 Fold Change | Std Error (log2) | p-Value | BY p-Value | Annotation |
|---|---|---|---|---|---|
| NET G1/G2 | |||||
| MME | −10.9 | 1.83 | 1.73 × 10−7 | 0.000178 | Cell functions, cell type-specific |
| ABCB1 | −5.3 | 0.968 | 9.42 × 10−7 | 0.000487 | CD molecules (MDR) |
| CASP1 | −4.23 | 0.725 | 2.41 × 10−7 | 0.000187 | Innate immune response |
| MAF | −3.87 | 0.923 | 9.14 × 10−5 | 0.0242 | Cell functions, cell type-specific, Th2 orientation |
| PSEN1 | −1.47 | 0.361 | 0.000132 | 0.0316 | T cell activation |
| CASP3 | −1.46 | 0.365 | 0.000181 | 0.0401 | Cell cycle checkpoint and arrest, co-regulators of autophagy and apoptosis/cell cycle, negative regulation of cell cycle |
| DOCK9 | 1.95 | 0.43 | 2.89 × 10−5 | 0.00995 | Cell functions, cell type-specific |
| TOLLIP | 2.33 | 0.498 | 1.74 × 10−5 | 0.00677 | Innate immune response |
| PNMA1 | 2.46 | 0.623 | 0.000216 | 0.0447 | Inflammatory response to antigenic stimulus |
| MAPKAPK2 | 2.5 | 0.495 | 4.45 × 10−6 | 0.00197 | Innate immune response |
| TFEB | 2.82 | 0.674 | 9.37 × 10−5 | 0.0242 | Humoral immune response |
| NT5E | 3.14 | 0.732 | 6.66 × 10−5 | 0.0207 | CD molecules, phagocytosis |
| IL1RAP | 4.25 | 0.774 | 8.96 × 10−7 | 0.000487 | Innate immune response |
| MNX1 | 5.59 | 0.821 | 5.75 × 10−9 | 1.78 × 10−5 | Humoral immune response |
| TNFRSF11B | 7.24 | 1.22 | 1.55 × 10−7 | 0.000178 | TNF superfamily members and their receptors |
| NET G3/NEC | |||||
| MME | −10 | 1.76 | 4.13 × 10−7 | 0.000427 | Cell functions, cell type-specific |
| ABCB1 | −5.49 | 0.925 | 1.66 × 10−7 | 0.000257 | CD molecules (MDR) |
| TPSAB1 | −5.11 | 1.27 | 0.00016 | 0.0496 | Cell functions, cell type-specific |
| MAF | −4.32 | 0.882 | 7.95 × 10−6 | 0.00617 | Cell functions, cell type-specific, Th2 orientation |
| CASP1 | −4.2 | 0.691 | 9.57 × 10−8 | 0.000257 | Innate immune response |
| AMICA1 | −2.84 | 0.699 | 0.000146 | 0.0496 | Regulation of immune response |
| MIF | 2.43 | 0.52 | 1.80 × 10−5 | 0.0111 | Innate immune response |
| DUSP4 | 4.61 | 1.01 | 2.70 × 10−5 | 0.014 | Innate immune response |
| TNFRSF11B | 4.73 | 1.16 | 0.000143 | 0.0496 | TNF superfamily members and their receptors |
| SPP1 | 5.89 | 1.43 | 0.000123 | 0.0496 | Cytokines and receptors |
| NET G3 (N) | NEC G3 (N) | |
|---|---|---|
| Patients | 18 | 11 |
| Stage II/III/IV/not known | 3/4/7/4 | 0/3/8/0 |
| Tissue samples | 20 | 13 |
| Primary tumor sites (N) | Gastrointestinal (3), bile duct (1), pancreas (7), liver (1), prostate (1), ovary (1), skin (1), kidney (2), CUP (1) | Gastrointestinal (7), bile duct (2), (bladder (1), CUP (1) |
| Patients with primary tumor/metastasis analyzed * | 18/2 | 11/1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busse, A.; Mochmann, L.H.; Spenke, C.; Arsenic, R.; Briest, F.; Jöhrens, K.; Lammert, H.; Sipos, B.; Kühl, A.A.; Wirtz, R.; et al. Immunoprofiling in Neuroendocrine Neoplasms Unveil Immunosuppressive Microenvironment. Cancers 2020, 12, 3448. https://doi.org/10.3390/cancers12113448
Busse A, Mochmann LH, Spenke C, Arsenic R, Briest F, Jöhrens K, Lammert H, Sipos B, Kühl AA, Wirtz R, et al. Immunoprofiling in Neuroendocrine Neoplasms Unveil Immunosuppressive Microenvironment. Cancers. 2020; 12(11):3448. https://doi.org/10.3390/cancers12113448
Chicago/Turabian StyleBusse, Antonia, Liliana H. Mochmann, Christiane Spenke, Ruza Arsenic, Franziska Briest, Korinna Jöhrens, Hedwig Lammert, Bence Sipos, Anja A. Kühl, Ralph Wirtz, and et al. 2020. "Immunoprofiling in Neuroendocrine Neoplasms Unveil Immunosuppressive Microenvironment" Cancers 12, no. 11: 3448. https://doi.org/10.3390/cancers12113448
APA StyleBusse, A., Mochmann, L. H., Spenke, C., Arsenic, R., Briest, F., Jöhrens, K., Lammert, H., Sipos, B., Kühl, A. A., Wirtz, R., Pavel, M., Hummel, M., Kaemmerer, D., Baum, R. P., & Grabowski, P. (2020). Immunoprofiling in Neuroendocrine Neoplasms Unveil Immunosuppressive Microenvironment. Cancers, 12(11), 3448. https://doi.org/10.3390/cancers12113448