CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults

 ,
,  ,
, 
Abstract
:1. B-Cell Acute Lymphoblastic Leukemia
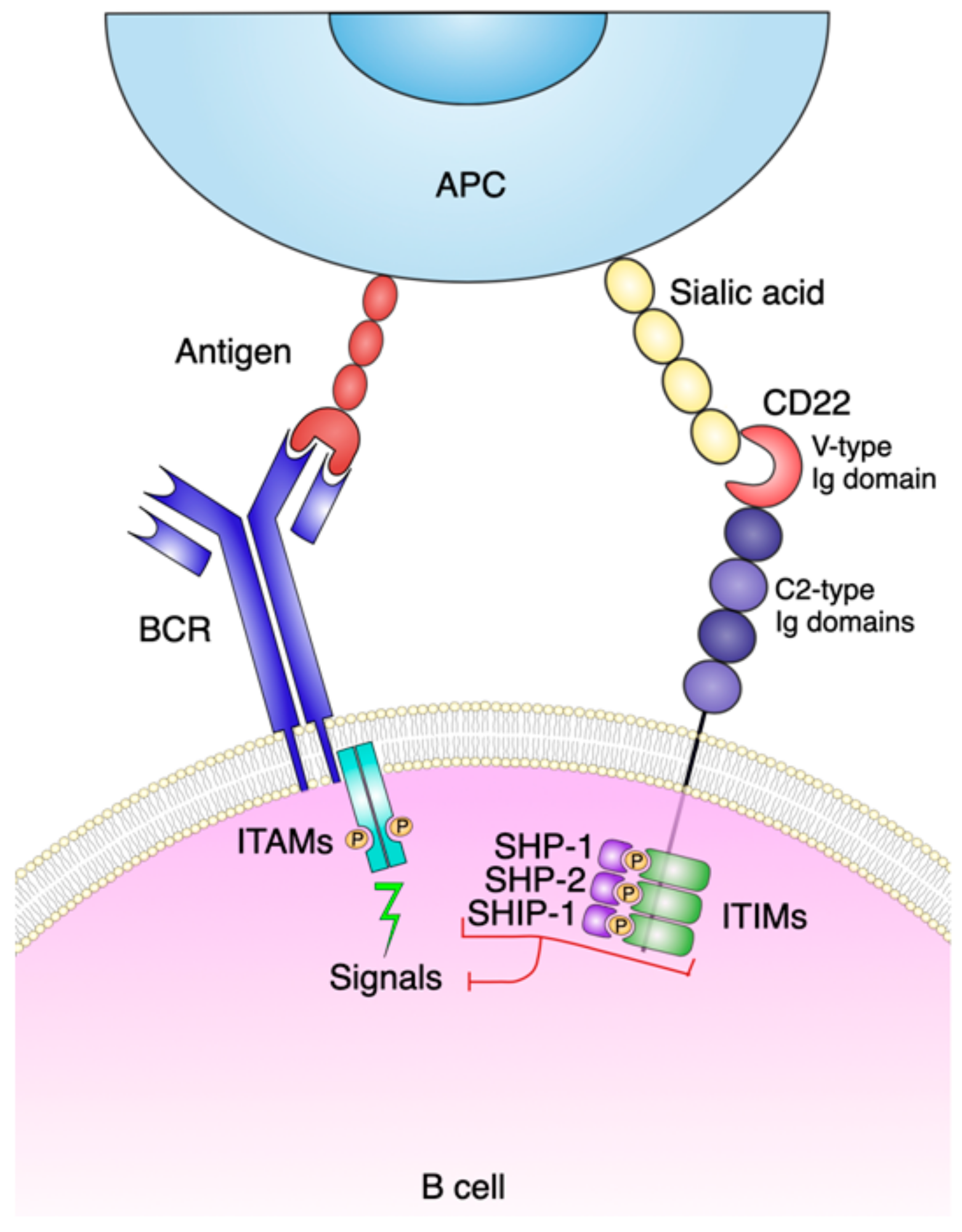
2. CD22 Molecule and Function
3. In Vivo Targeting of Human CD22
4. Therapeutic Anti-CD22 Antibodies in B-ALL
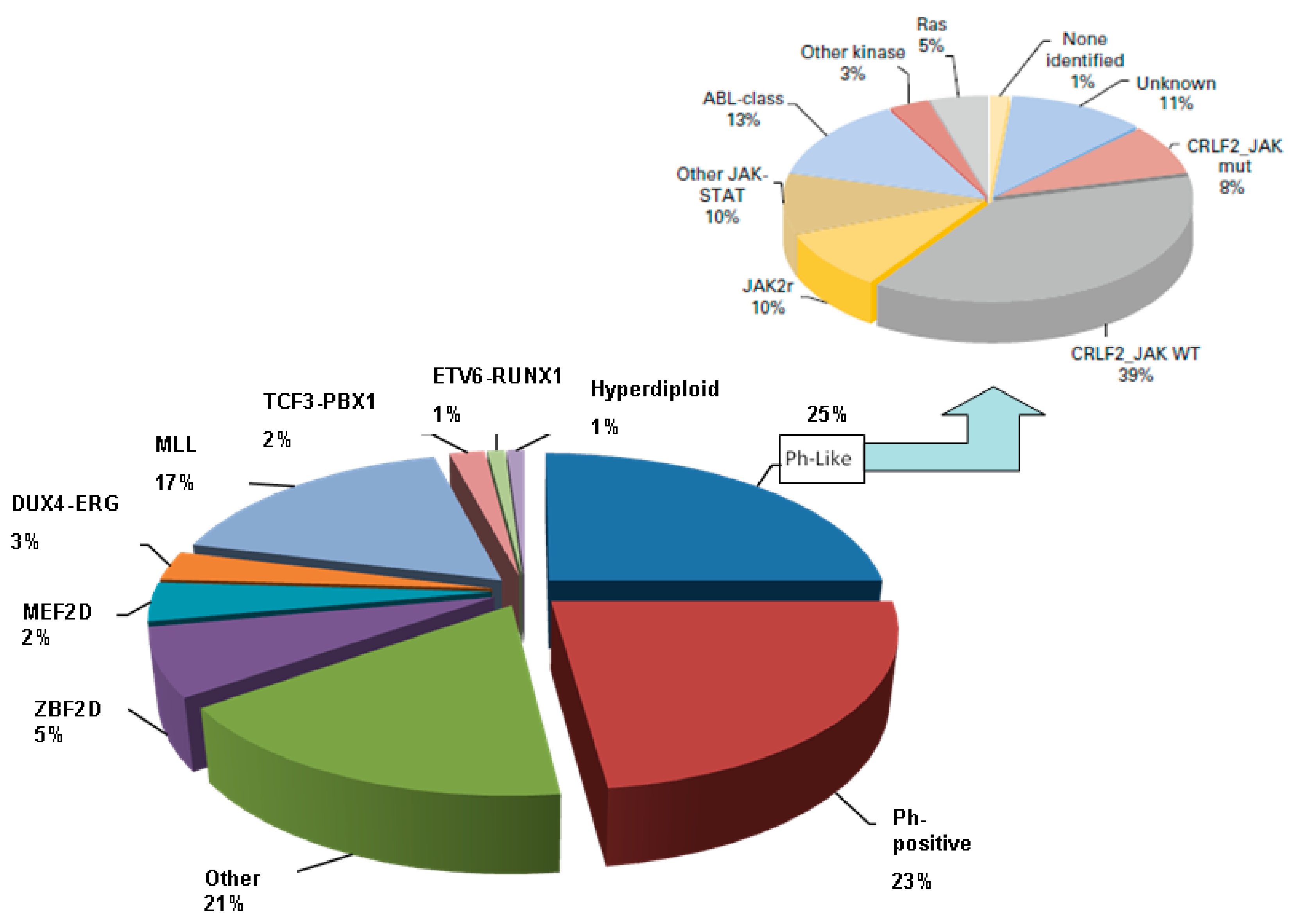
5. B-ALL Subgroups
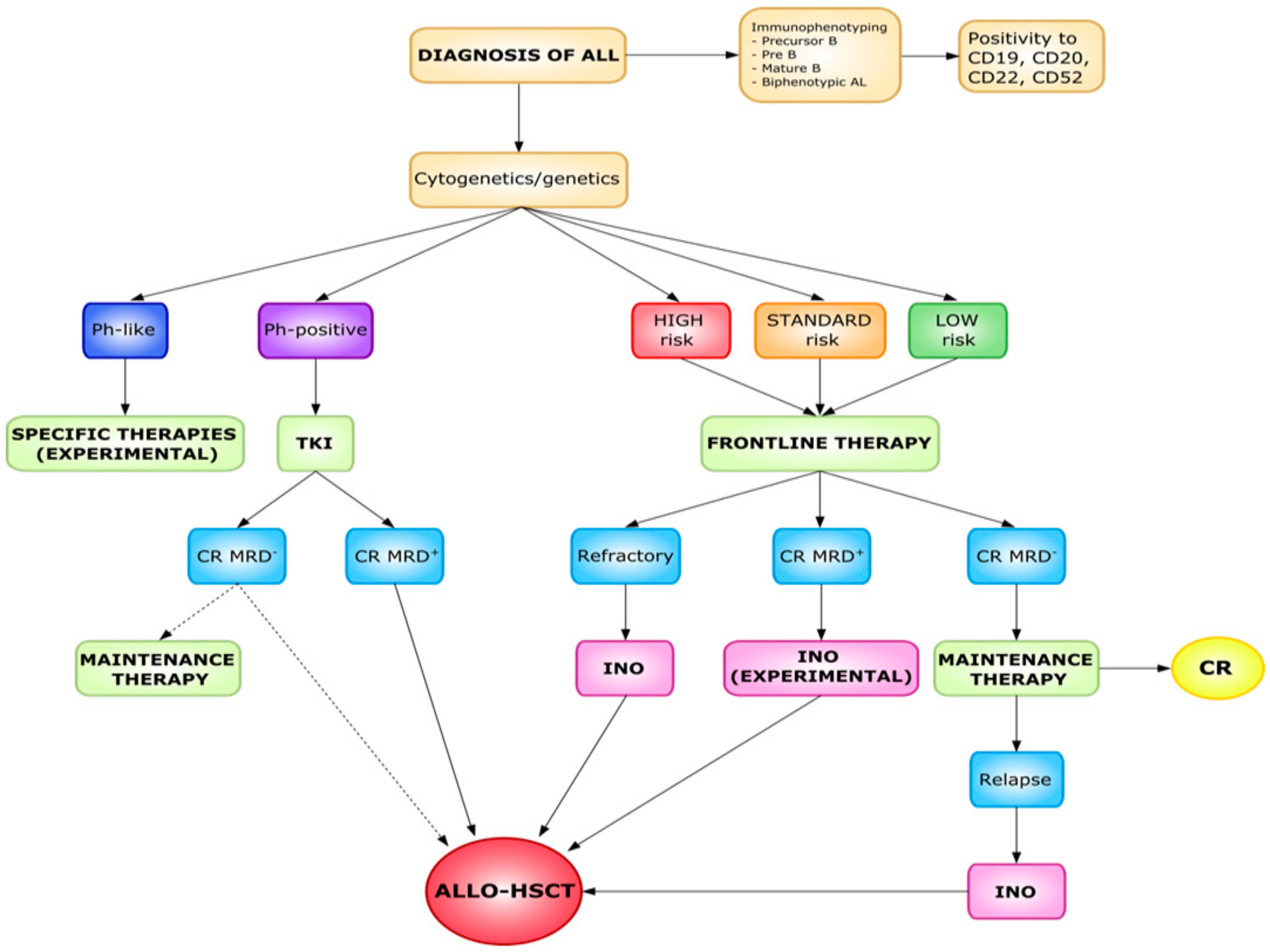
6. Predictors of Response in ALL
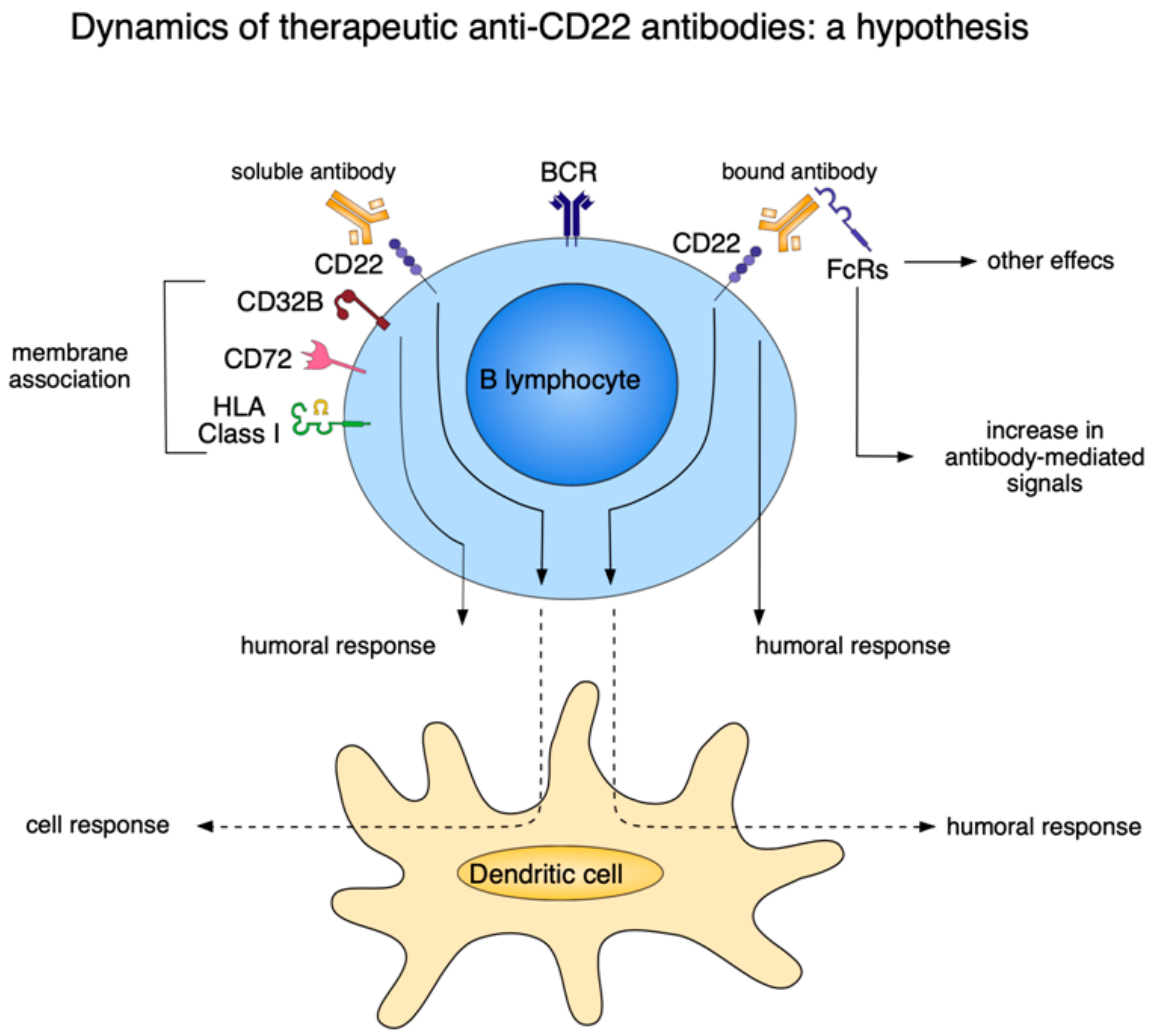
7. What Can Basic Science Add to Antibody Therapy
8. Alternative Approaches: CD22 CAR T-Cell Therapy in Refractory or Relapsed B-ALL
9. Concluding Remarks
Funding
Conflicts of Interest
References
- Jabbour, E.; O’Brien, S.; Konopleva, M.; Kantarjian, H. New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer 2015, 121, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Maffini, E.; Saraceni, F.; Lanza, F. Treatment of Adult Patients with Relapsed/Refractory B-Cell Philadelphia-Negative Acute Lymphoblastic Leukemia. Clin. Hematol. Int. 2019, 1, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, I.; Mulligan, C.G. Genetic basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Frisch, A.; Ofran, Y. How I diagnose and manage Philadelphia chromosome-like acute lymphoblastic leukemia. Haematologica 2019, 104, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; De Angelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Dinner, S.; Liedtke, M. Antibody-based therapies in patients with acute lymphoblastic leukemia. Hematology Am. Soc. Hematol. Educ. Program 2018, 1, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Lanza, F. Issue Highlight—July 2018. Cytometry 2018, 94, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Poe, J.C.; Fujimoto, Y.; Hasegawa, M.; Haas, K.M.; Miller, A.S.; Sanford, I.G.; Bock, C.B.; Fujimoto, M.; Tedder, T.F. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat. Immunol. 2004, 5, 1078–1087. [Google Scholar] [CrossRef]
- Moldenhawer, C.; Doerken, B.; Schwartz, R.; Pezzutto, A.; Hammerling, G.J. Analysis of Ten B Lymphocyte-Specific Workshop Monoclonal Antibodies. In Leukocytes Typing II; Reinherz, A., Haynes, B.F., Eds.; Springer-Verlag: New York, NY, USA, 1986; Volume 2, pp. 61–67. ISBN 978-1-4612-4848-4. [Google Scholar]
- Meyer, S.J.; Linder, A.T.; Brandl, C.; Nitschke, L. B Cell Siglecs-News on Signaling and Its Interplay With Ligand Binding. Front Immunol. 2018, 2820, 3–9. [Google Scholar] [CrossRef]
- Poe, J.C.; Tedder, T.F. CD22 and Siglec-G in B cell function and tolerance. Trends Immunol. 2012, 33, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedder, T.F.; Poe, J.C.; Haas, K.M. CD22: A multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv. Immunol. 2005, 88, 1–50. [Google Scholar] [PubMed]
- Collins, B.E.; Smith, B.A.; Bengtson, P.; Paulson, J.C. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat. Immunol. 2006, 7, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Wang, T.T.; Dahan, R.; Maamary, J.; Ravetch, J.V. Signaling by Antibodies: Recent Progress. Annu. Rev. Immunol. 2017, 35, 285–311. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Harumiya, S.; Takematsu, H.; Kozutsumi, Y.; Wabl, M.; Fujimoto, M.; Tedder, T.F. CD22 serves as a receptor for soluble IgM. Eur. J. Immunol. 2012, 42, 241–247. [Google Scholar] [CrossRef]
- Moek, K.L.; de Groot, D.J.; de Vries, E.G.; Fehrmann, R.S. The antibody-drug conjugate target landscape across a broad range of tumour types. Ann. Oncol. 2017, 28, 3083–3091. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Sicard, T.; Cui, H.; Mazhab-Jafari, M.T.; Benlekbir, S.; Guarné, A.; Rubinstein, J.L.; Julien, J.P. Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 2017, 8, 764. [Google Scholar]
- Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zito, A.; Mariani, V.; Faini, A.C.; Morandi, F.; Schiavoni, I.; Ausiello, C.M.; Malavasi, F. Antibody mimicry, receptors and clinical applications. Hum. Antibodies 2017, 25, 75–85. [Google Scholar] [CrossRef]
- Walker, J.A.; Smith, K.G. Dependence of surface monoclonal antibody binding on dynamic changes in Fc gamma RIIb expression. Immunology 2008, 124, 412–418, Erratum in: Immunology 2008, 124, 417. [Google Scholar] [CrossRef]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Bolzoni, M.; Toscani, D.; Costa, F.; Castella, B.; Faini, A.C.; Massaia, M.; Pistoia, V.; et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD. Oncoimmunology 2018, 7, e1574198. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Heiser, R.A.; Detanico, T.O.; Getahun, A.; Kirchenbaum, G.A.; Casper, T.L.; Aydintug, M.K.; Carding, S.R.; Ikuta, K.; Huang, H.; et al. γδ T cells affect IL-4 production and B-cell tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, E39–E48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudge, E.U.; Cutler, A.J.; Pritchard, N.R.; Smith, K.G. Interleukin 4 reduces expression of inhibitory receptors on B cells and abolishes CD22 and Fc gamma RII-mediated B cell suppression. J. Exp. Med. 2002, 195, 1079–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raponi, S.; De Propris, M.S.; Intoppa, S.; Milani, M.L.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foá, R.; Guarini, A. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: Analysis of 552 cases. Leuk. Lymphoma 2011, 52, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- De Angelo, D.J.; Stock, W.; Stein, A.S.; Shustov, A.; Liedtke, M.; Schiffer, C.A.; Vandendries, E.; Liau, K.; Ananthakrishnan, R.; Boni, J.; et al. Inotuzumab ozogamicin in adults with relapsed or refractory CD22-positive acute lymphoblastic leukemia: A phase 1/2 study. Blood Adv. 2017, 1, 1167–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From preclinical development to success in B-cell acute lymphoblastic leukemia. Blood Adv. 2019, 3, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: Results of a phase I study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Yurkiewicz, I.R.; Muffly, L.; Liedtke, M. Inotuzumab ozogamicin: A CD22 mAb-drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug. Des. Devel. Ther. 2018, 12, 2293–2300. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.B.; Gooley, T.A.; van der Velden, V.H.; Loken, M.R.; van Dongen, J.J.; Flowers, D.A.; Bernstein, I.D.; Appelbaum, F.R. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007, 109, 4168–4170. [Google Scholar] [CrossRef]
- Cianfriglia, M.; Mallano, A.; Ascione, A.; Dupuis, M.L. Multidrug transporter proteins and cellular factors involved in free and mAb linked calicheamicin-gamma1 (gentuzumab ozogamicin, GO) resistance and in the selection of GO resistant variants of the HL60 AML cell line. Int. J. Oncol. 2010, 36, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, R.; Chauhan, L.; Alonzo, T.A.; Wang, Y.C.; Elmasry, A.; Loken, M.R.; Pollard, J.; Aplenc, R.; Raimondi, S.; Hirsch, B.A.; et al. ABCB1 SNP predicts outcome in patients with acute myeloid leukemia treated with Gemtuzumab ozogamicin: A report from Children’s Oncology Group AAML0531 Trial. Blood Cancer J. 2019, 9, 51. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; De Angelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; White, J.L.; et al. Inotuzumab Ozogamicin Versus Standard of Care in Relapsed or Refractory Acute Lymphoblastic Leukemia: Final Report and Long-Term Survival Follow-Up From the Randomized, Phase 3 INO-VATE Study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Ravandi, F.; Kebriaei, P.; Huang, X.; Short, N.J.; Thomas, D.; Sasaki, K.; Rytting, M.; Jain, N.; Konopleva, M.; et al. Salvage chemoimmunotherapy with inotuzumab ozogamicin combined with mini–hyper-CVD for patients with relapsed or refractory philadelphia chromosome–negative acute lymphoblastic leukemia. JAMA Oncol. 2018, 4, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.J.; De Angelo, D.J.; Stelljes, M.; Stock, W.; Liedtke, M.; Gökbuget, N.; O’Brien, S.M.; Wang, T.; Paccagnella, M.L.; Sleight, B.; et al. Efficacy and Safety Analysis by Age Cohort of Inotuzumab Ozogamicin in Patients With Relapsed or Refractory Acute Lymphoblastic Leukemia Enrolled in INO-VATE. Cancer 2018, 124, 1722–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebriaei, P.; Cutler, C.; de Lima, M.; Giralt, S.; Lee, S.J.; Marks, D.; Merchant, A.; Stock, W.; van Besien, K.; Stelljes, M. Management of important adverse events associated with inotuzumab ozogamicin: Expert panel review. Bone Marrow Transplant. 2018, 53, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Advani, A.S.; Stelljes, M.; Kebriaei, P.; Cassaday, R.D. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: Results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017, 4, e387–e398. [Google Scholar] [CrossRef]
- Mohty, M.; Malard, F.; Abecasis, M.; Aerts, E.; Alaskar, A.S.; Aljurf, M.; Arat, M.; Bader, P.; Baron, F.; Basak, G.; et al. Prophylactic, preemptive, and curative treatment for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: A position statement from an international expert group. Bone Marrow Transplant. 2019. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Su, Y.; Jabbour, E.J.; Bhattacharyya, H.; Yan, E.; Cappelleri, J.C.; Marks, D.I. Patient-Reported Outcomes From a Phase 3 Randomized Controlled Trial of Inotuzumab Ozogamicin Versus Standard Therapy for Relapsed/Refractory Acute Lymphoblastic Leukemia. Cancer 2018, 124, 2151–2160. [Google Scholar] [CrossRef]
- Advani, A.S.; Moseley, A.; Liedtke, M.; O’Donnell, M.R.; Aldoss, I.; Mims, M.P.; O’Dwyer, K.M.; Othus, M.; Erba, H.P. SWOG 1312 Final Results: A Phase 1 Trial of Inotuzumab in Combination with CVP (Cyclophosphamide, Vincristine, Prednisone) for Relapsed/Refractory CD22+ Acute Leukemia. Blood 2019, 134, 227. [Google Scholar] [CrossRef]
- Yang, X.; King, A.C.; Kabel, C.C.; Forlenza, C.J.; Park, J.H.; Geyer, M.B. Inotuzumab Ozogamicin Is An Effective Salvage Therapy in Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia with High-Risk Molecular Features, Including TP53 Loss. Blood 2019, 134, 3888. [Google Scholar] [CrossRef]
- Cherian, S.; Miller, V.; McCullouch, V.; Dougherty, K.; Fromm, J.R.; Wood, B.L. A Novel Flow Cytometric Assay for Detection of Residual Disease in Patients with B-Lymphoblastic Leukemia/Lymphoma Post Anti-CD19 Therapy. Cytometry Part B 2018, 94B, 112–120. [Google Scholar] [CrossRef]
- Rank, C.U.; Stock, W. Should immunologic strategies be incorporated into frontline ALL therapy? Best Pract. Res. Clin. Haematol. 2018, 31, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Di Lillo, D.J.; Ravetch, J.V. Fc-Receptor Interactions Regulate Both Cytotoxic and Immunomodulatory Therapeutic Antibody Effector Functions. Cancer Immunol. Res. 2015, 3, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarth, P.M.; Pietersz, G.A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 2012, 11, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Faini, A.C. Mechanism of Action of a New Anti-CD38 Antibody: Enhancing Myeloma Immunotherapy. Clin. Cancer Res. 2019, 25, 2946–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyzik, M.; Rath, T.; Lencer, W.I.; Baker, K.; Blumberg, R.S. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J. Immunol. 2015, 194, 4595–4603. [Google Scholar] [CrossRef] [Green Version]
- Van de Donk, N.W.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Farrokhi, V.; Caiazzo, T.; Wang, M.; O’Hara, D.M.; Neubert, H. Human FcRn Tissue Expression Profile and Half-Life in PBMCs. Biomolecules 2019, 9, 373. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.M.; Zhang, Z.; Jasper, P.; Luo, H.; Avery, L.B.; King, L.E.; Neubert, H.; Barton, H.A.; Betts, A.M.; Webster, R. A Physiologically-Based Pharmacokinetic Model for the Prediction of Monoclonal Antibody Pharmacokinetics from In Vitro Data. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, H.M.; Zhu, A.Z.X. A minimal physiologically-based pharmacokinetic model demonstrates role of FcRn competition in drug-disease interactions with antibody therapy. Clin. Pharmacol. Ther. 2020, 107, 423–434. [Google Scholar] [CrossRef]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019, 33, 2854–2866. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, R. Advances in off-the-shelf CAR T-cell therapy. Clin. Adv. Hematol Oncol. 2019, 17, 155–157. [Google Scholar] [PubMed]
- Atanackovic, D.; Yousef, S.; Shorter, C.; Tantravahi, S.K.; Steinbach, M.; Iglesias, F.; Sborov, D.; Radhakrishnan, S.V.; Chiron, M.; Miles, R.; et al. In vivo vaccination effect in multiple myeloma patients treated with the monoclonal antibody isatuximab. Leukemia 2020, 34, 317–321. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Phase | Disease | Intervention | Results | Reference |
|---|---|---|---|---|
| 2 | R/R Ph-Negative CD22 positive ALL | Mini-Hyper-CVD combined with InO and Rituximab | ORR was 78% (59% CR) MRD negative rates of 52% (at time of morphological response) and 82% (at three months). Median RFS of 8 months. Median OS of 11 months | [34] |
| 3 | R/R ALL | 0.8 mg/m2 (D1), 0.5 mg/m2 (D8), 0.5 mg/m2 (D15) Versus Standard therapy | CR + CRi 80.7% (CR 35.8%). Median RFS of five months. Median OS of 7.7 months | [6] |
| 1/2 | R/R ALL | 1.8 mg/m2 weekly | 69% CR/CRi (29% CR) | [25] |
| 2 | B-ALL with positive MRD | InO | Recruiting | NCT03441061 |
| 4 | R/R B-ALL | Investigating InO lower dose level (1.2 mg/m2/cycle) for those with higher risk for liver toxicity or VOD | Recruiting | NCT03677596 |
| 2 | Precursor B-cell ALL in 56–74 years old | InO induction followed by conventional chemotherapy | Recruiting | NCT03460522 |
| 1/2 | Ph + B-ALL and CML-blast phase | Bosutinib plus InO | Recruiting | NCT02311998 |
| 1 | Acute leukemia of ambiguous lineage, Recurrent Ph + B-ALL, Recurrent Burkitt Lymphoma | Inotuzumab plus CVP (cyclophosphamide, Vincristine, Prednisone) | Recruiting | NCT01925131 |
| 2 | Ph negative B-ALL | Inotuzumab followed by Blinatumomab | Recruiting | NCT03739814 |
| 1/2 | R/R B-ALL | Inotuzumab plus Vincristine (liposomal) | Not yet recruiting | NCT03851081 |
| 2 | Ph negative B-ALL in 55 years or older | InO plus CVP induction | Recruiting | NCT03249870 |
| 2 | ALL with positive MRD prior to HSCT | InO | Not yet recruiting | NCT03610438 |
| 1/2 | Untreated ALL in 60 years and older | InO plus combination chemotherapy | Recruiting | NCT01371630 |
| 3 | Newly diagnosed B-ALL in 18–39 years old | InO plus chemotherapy | Recruiting | NCT03150693 |
| 2 | R/R ALL | Lower dose InO | Recruiting | NCT03094611 |
| 2 | ALL | InO plus Hyper-CVAD | Recruiting | NCT03488225 |
| 2 | B-ALL in 1–21 years old | InO | Recruiting | NCT02981628 |
| 3 | ALL | Tisagenlecleucel versus Blinatumomab or Inotuzumab | Not yet recruiting | NCT03628053 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanza, F.; Maffini, E.; Rondoni, M.; Massari, E.; Faini, A.C.; Malavasi, F. CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers 2020, 12, 303. https://doi.org/10.3390/cancers12020303
Lanza F, Maffini E, Rondoni M, Massari E, Faini AC, Malavasi F. CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers. 2020; 12(2):303. https://doi.org/10.3390/cancers12020303
Chicago/Turabian StyleLanza, Francesco, Enrico Maffini, Michela Rondoni, Evita Massari, Angelo Corso Faini, and Fabio Malavasi. 2020. "CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults" Cancers 12, no. 2: 303. https://doi.org/10.3390/cancers12020303
APA StyleLanza, F., Maffini, E., Rondoni, M., Massari, E., Faini, A. C., & Malavasi, F. (2020). CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers, 12(2), 303. https://doi.org/10.3390/cancers12020303