Cinnamaldehyde Enhances Antimelanoma Activity through Covalently Binding ENO1 and Exhibits a Promoting Effect with Dacarbazine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
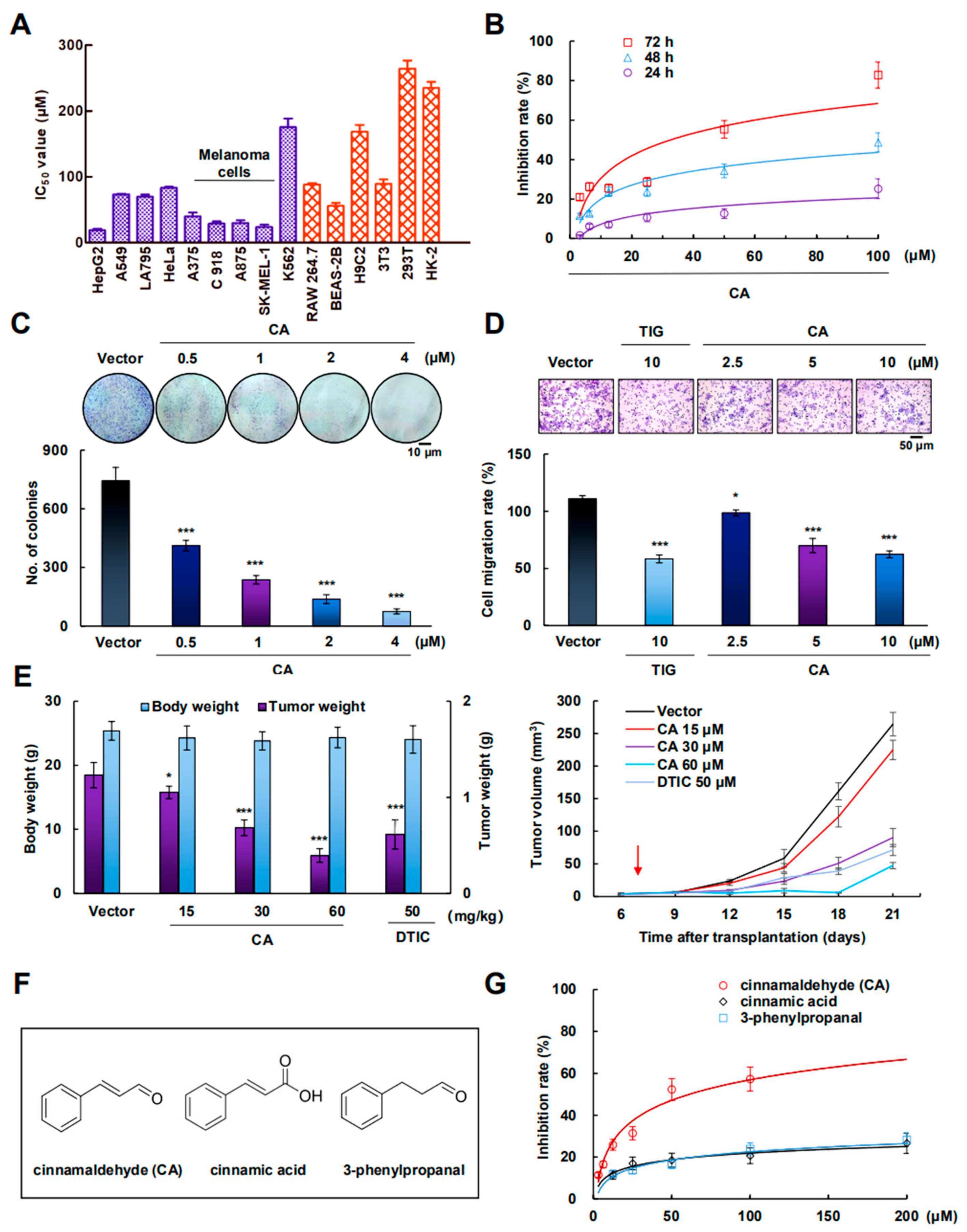
2.1. CA Inhibited A375 Cell Growth In Vitro and In Vivo
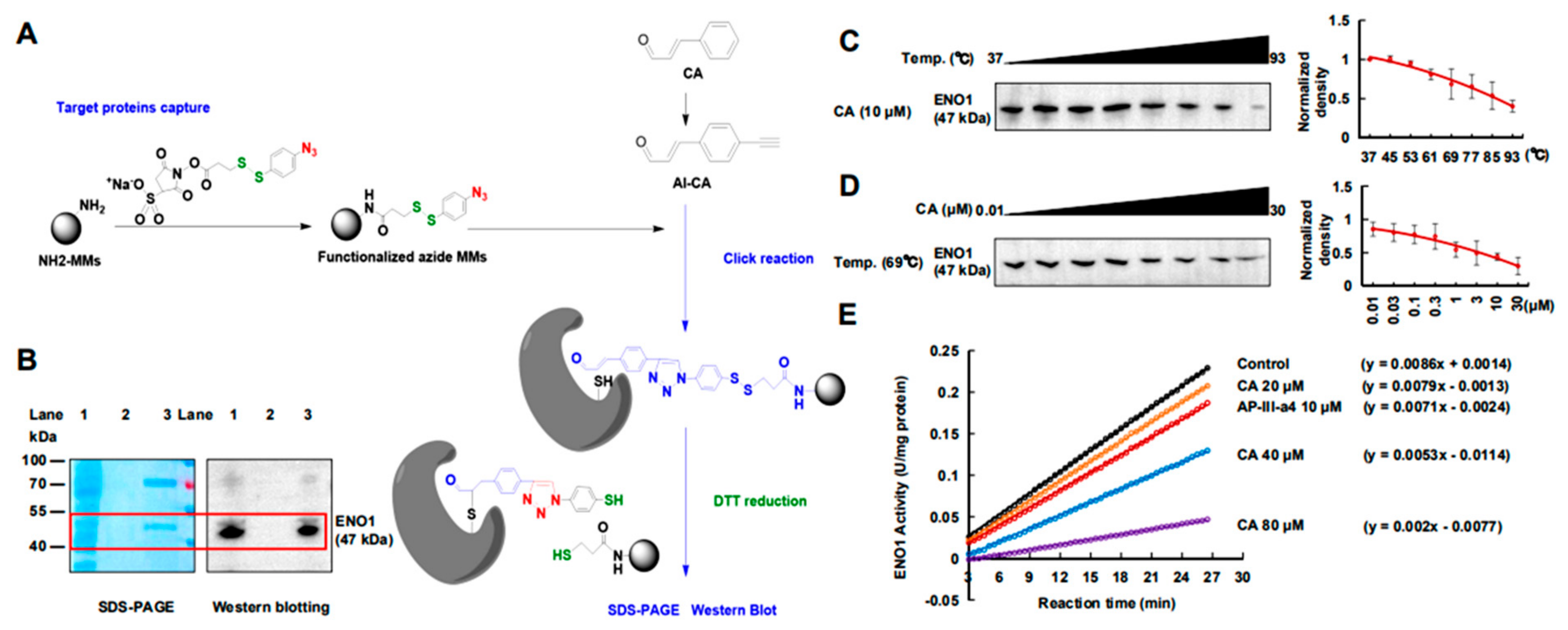
2.2. CA Covalently Bonded to ENO1 Protein
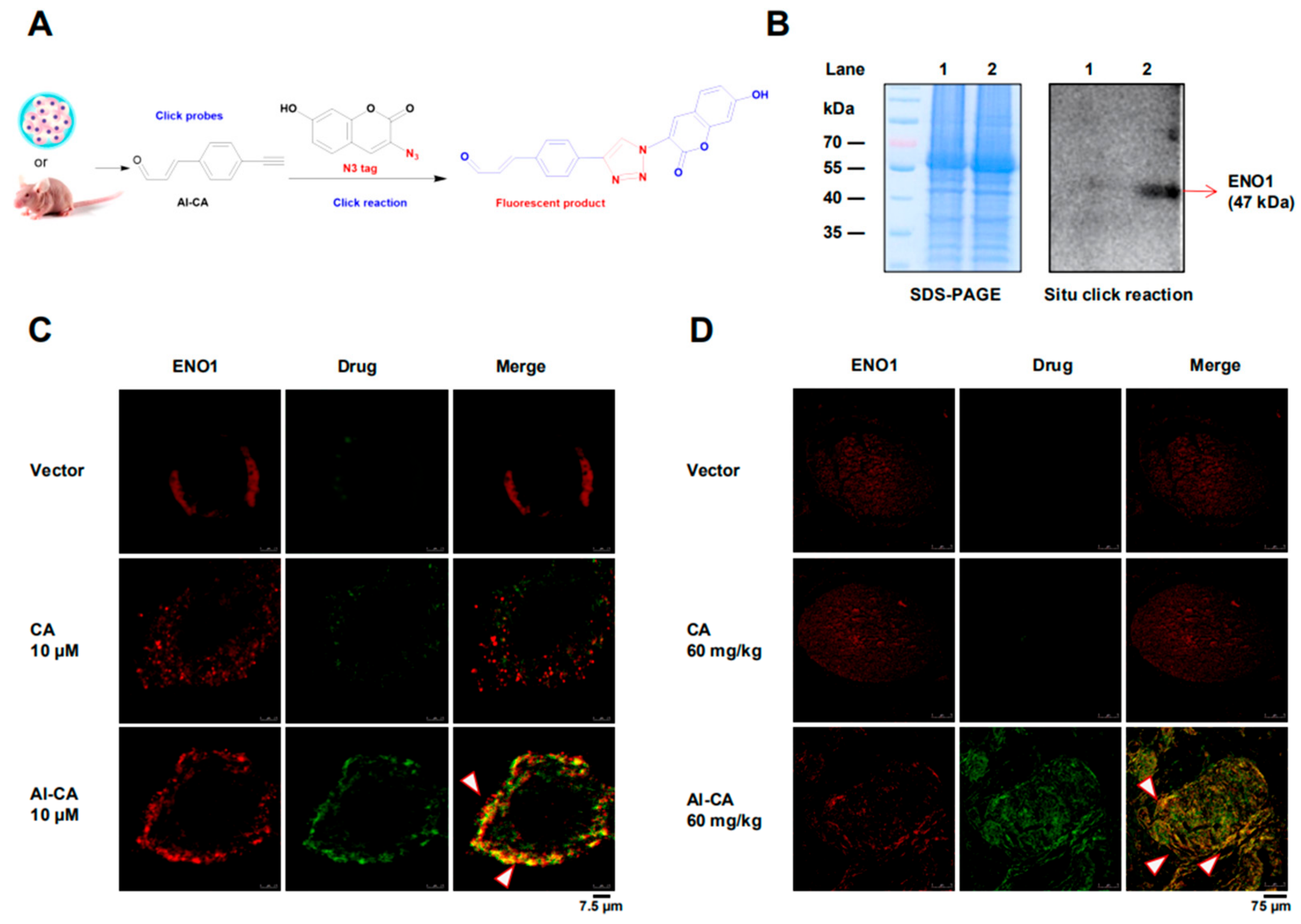
2.3. CA Colocalized with ENO1 in both A375 Cells and Living Tumors
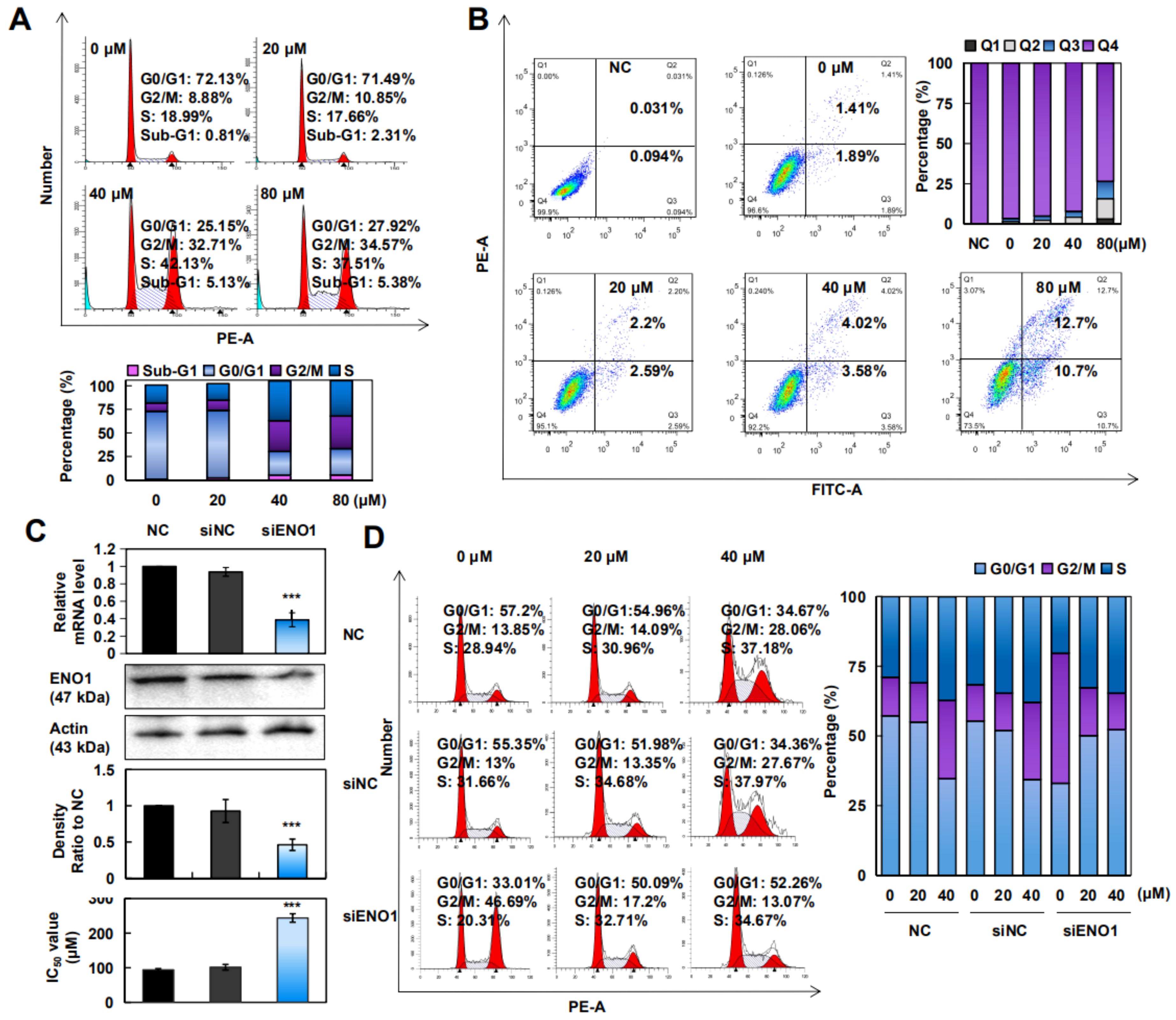
2.4. CA Induced Apoptosis and Arrested the G2/M/S Phase in A375 Cells
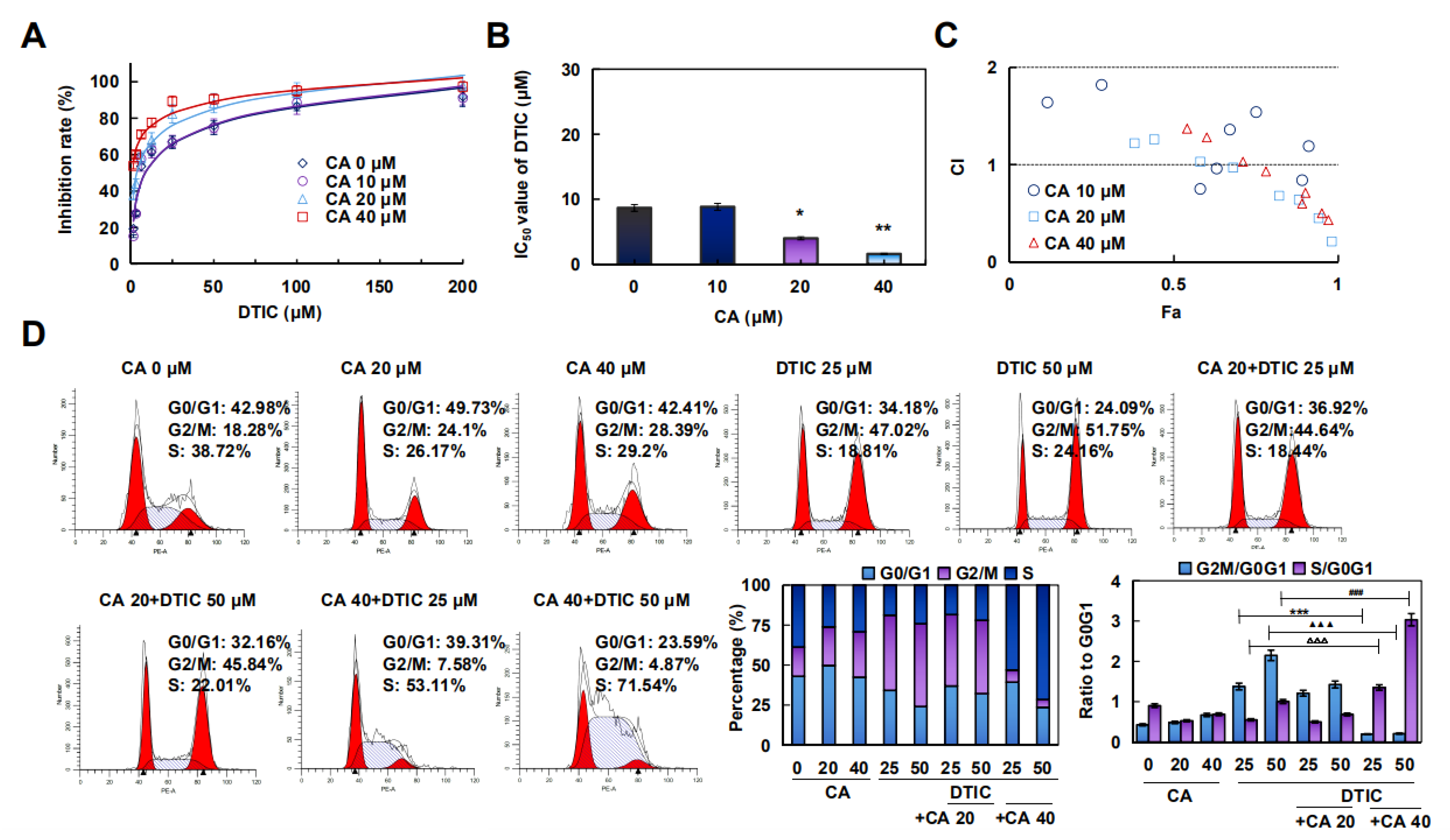
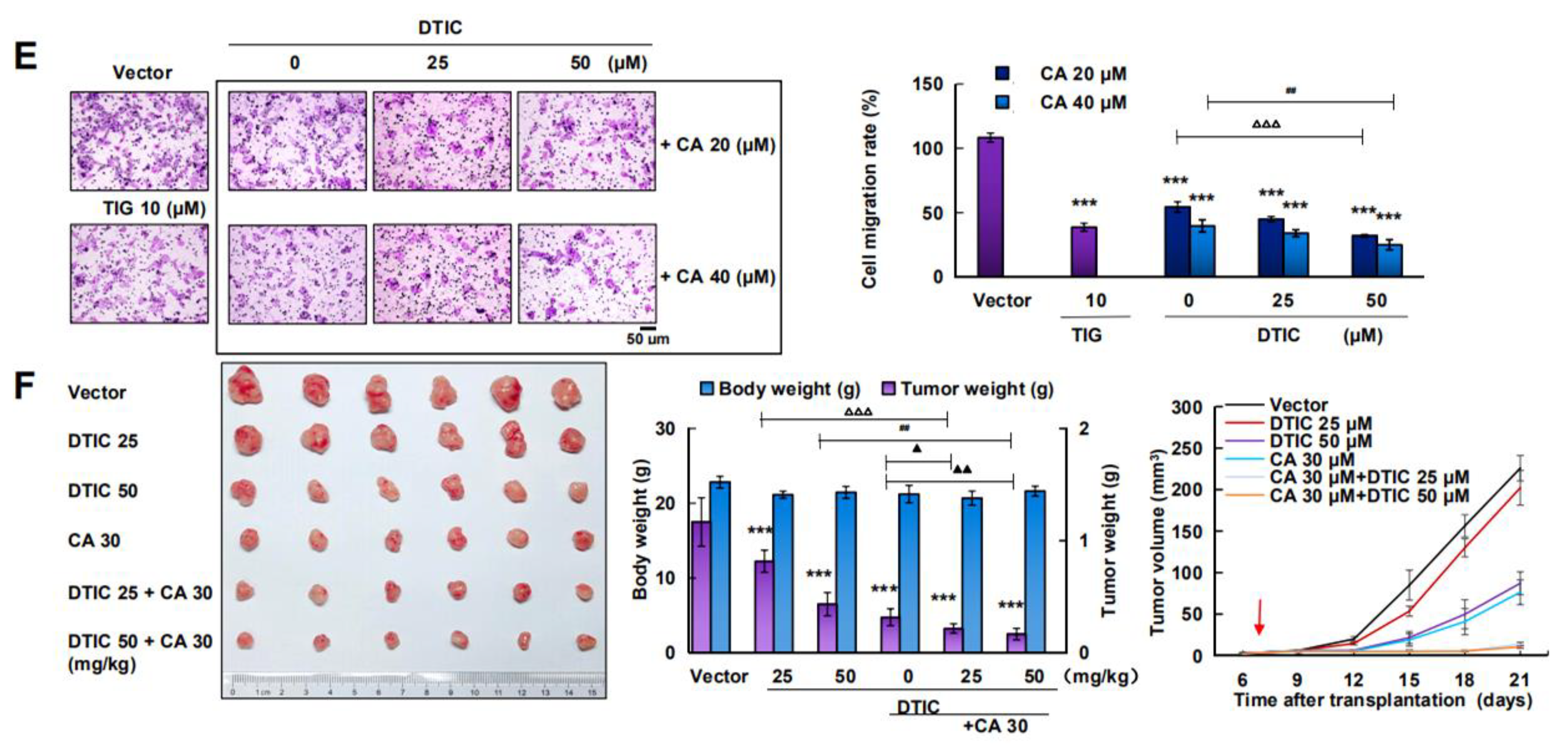
2.5. CA Combined with DTIC Promoted Antimelanoma Effect In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Cell Viability Assessment
4.3. Colony Formation and Cell Migration Assays
4.4. Cell Cycle and Annexin V-FITC/PI Double-Staining Assays
4.5. Enrichment of Target and Western Blotting Assay
4.6. Cellular Thermal Shift Assay (CETSA)
4.7. Colocalization of Target Proteins with AL-CA Probe
4.8. RNA Interference and Quantitative Real-Time PCR (RT-PCR) Analysis
4.9. Xenografts In Vivo
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. CA: A cancer journal for clinicians. Cancer Stat. 2017, 67, 7–30. [Google Scholar]
- Zhou, L.; Lu, Y.; Yang, G.; Wu, J. Research on tumorigenicity of cinnamaldehyde in melanoma cell lines and its mechanism. Tumour Biol. 2014, 35, 5717–5722. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Chen, Y.S.; Liu, F.; Luo, Y.H.; Fan, Y.; Xu, F.G.; Li, P.; Zhou, B.; Pan, X.Y.; Wang, C.C.; Cui, L. EDNRB isoform 3 confers Temozolomide resistance in A375 melanoma cells by modulating membrane potential, reactive oxygen species and mitochondrial Ca2+. Cancer Manag. Res. 2019, 11, 7353–7367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; McClay, E.F. Systemic chemotherapy for the treatment of metastatic melanoma. Semin. Oncol. 2002, 29, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Lillehammer, T.; Engesaeter, B.O.; Prasmickaite, L.; Maelandsmo, G.M.; Fodstad, O.; Engebraaten, O. Combined treatment with Ad-hTRAIL and DTIC or SAHA is associated with increased mitochondrial-mediated apoptosis in human melanoma cell lines. J. Gene Med. 2007, 9, 440–451. [Google Scholar] [CrossRef]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Nguyen, J.; Jhaveri, N.; Rosenstein-Sisson, R.; Hofman, F.M.; Schonthal, A.H. A novel temozolomide analog, NEO212, with enhanced activity against MGMT-positive melanoma in vitro and in vivo. Cancer Lett. 2015, 358, 144–151. [Google Scholar] [CrossRef]
- Loo, T.L.; Housholder, G.E.; Gerulath, A.H.; Saunders, P.H.; Farquhar, D. Mechanism of action and pharmacology studies with DTIC (NSC-45388). Cancer Treat. Rep. 1976, 60, 149–152. [Google Scholar]
- Beaumont, K.A.; Hill, D.S.; Daignault, S.M.; Lui, G.Y.L.; Sharp, D.M.; Gabrielli, B.; Weninger, W.; Haass, N.K. Cell Cycle Phase-Specific Drug Resistance as an Escape Mechanism of Melanoma Cells. J. Investig. Dermatol. 2016, 136, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Grossman, D.; Altieri, D.C. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev. 2001, 20, 3–11. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Obata, N.; Kawashima, K.; Tabata, M.; Imano, M.; Satou, T.; Nishida, S. Combination therapy with dacarbazine and statins improved the survival rate in mice with metastatic melanoma. J. Cell. Physiol. 2019, 234, 17975–17989. [Google Scholar] [CrossRef] [PubMed]
- Gander, M.; Leyvraz, S.; Decosterd, L.; Bonfanti, M.; Marzolini, C.; Shen, F.; Lienard, D.; Perey, L.; Colella, G.; Biollaz, J.; et al. Sequential administration of temozolomide and fotemustine: depletion of O6-alkyl guanine-DNA transferase in blood lymphocytes and in tumours. Ann. Oncol. 1999, 10, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Camlica, H.; Topuz, E. Temozolomide in combination with fotemustine in patients with metastatic melanoma. Cancer Chemother. Pharmacol. 2008, 62, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.C.; Roos, W.P.; Jost, E.; Belohlavek, C.; Lennerz, V.; Schmidt, C.W.; Christmann, M.; Kaina, B. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br. J. Cancer 2009, 100, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, H.; Liu, C.; Wang, L.; Ma, R.; Chen, B.; Li, L.; Niu, J.; Fu, M.; Zhang, D.; et al. Cinnamaldehyde in diabetes: A review of pharmacology, pharmacokinetics and safety. Pharmacol. Res. 2017, 122, 78–89. [Google Scholar] [CrossRef]
- Chen, B.J.; Fu, C.S.; Li, G.H.; Wang, X.N.; Lou, H.X.; Ren, D.M.; Shen, T. Cinnamaldehyde Analogues as Potential Therapeutic Agents. Mini Rev. Med. Chem. 2017, 17, 33–43. [Google Scholar] [CrossRef]
- Jiang, J.; Emont, M.P.; Jun, H.; Qiao, X.; Liao, J.; Kim, D.I.; Wu, J. Cinnamaldehyde induces fat cell-autonomous thermogenesis and metabolic reprogramming. Metabolism 2017, 77, 58–64. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Sun, J.; Wang, S. Protective effects of cinnamic acid and cinnamic aldehyde on isoproterenol-induced acute myocardial ischemia in rats. J. Ethnopharmacol. 2013, 150, 125–130. [Google Scholar] [CrossRef]
- Kurokawa, M.; Kumeda, C.A.; Yamamura, J.; Kamiyama, T.; Shiraki, K. Antipyretic activity of cinnamyl derivatives and related compounds in influenza virus-infected mice. Eur. J. Pharmacol. 1998, 348, 45–51. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, C.H.; Kim, M.S.; Kim, J.Y.; Jung, K.J.; Chung, J.H.; An, W.G.; Lee, J.W.; Yu, B.P.; Chung, H.Y. Suppression of age-related inflammatory NF-kappaB activation by cinnamaldehyde. Biogerontology 2007, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Ismail, I.A.; Kang, S.M.; Han, D.C.; Kwon, B.M. Cinnamaldehydes in Cancer Chemotherapy. Phytother. Res. 2016, 30, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Geng, M.; Du, Z.; Yao, J.; Zheng, Y.; Li, Z.; Zhang, Z.; Li, J.; Duan, Y.; Du, G. Berberine and cinnamaldehyde together prevent lung carcinogenesis. Oncotarget 2017, 8, 76385–76397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Obianom, O.N.; Ai, Y. Novel cinnamaldehyde-based aspirin derivatives for the treatment of colorectal cancer. Bioorg. Med. Chem. Lett. 2018, 28, 2869–2874. [Google Scholar] [CrossRef]
- Wani, K.D.; Kadu, B.S.; Mansara, P.; Gupta, P.; Deore, A.V.; Chikate, R.C.; Poddar, P.; Dhole, S.D.; Kaul-Ghanekar, R. Synthesis, characterization and in vitro study of biocompatible cinnamaldehyde functionalized magnetite nanoparticles (CPGF Nps) for hyperthermia and drug delivery applications in breast cancer. PLoS ONE 2014, 9, e107315. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B.; Lamore, S.D.; Ley, S.; Bause, A.S.; Azimian, S.; Wondrak, G.T. The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth. Free Radic. Biol. Med. 2009, 46, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wu, J.; Li, X.; Liu, H.; Qin, J.; Bai, Z.; Chi, B.; Chen, X. Identification and validation nucleolin as a target of curcumol in nasopharyngeal carcinoma cells. J. Proteomics 2018, 182, 1–11. [Google Scholar] [CrossRef]
- Liu, C.X.; Yin, Q.Q.; Zhou, H.C.; Wu, Y.L.; Pu, J.X.; Xia, L.; Liu, W.; Huang, X.; Jiang, T.; Wu, M.X.; et al. Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 2012, 8, 486–493. [Google Scholar] [CrossRef]
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef]
- Gersch, M.; Kreuzer, J.; Sieber, S.A. Electrophilic natural products and their biological targets. Nat. Product Rep. 2012, 29, 659–682. [Google Scholar] [CrossRef]
- Houghton, A.N.; Real, F.X.; Davis, L.J.; Cordon-Cardo, C.; Old, L.J. Phenotypic heterogeneity of melanoma. Relation to the differentiation program of melanoma cells. J. Exp. Med. 1987, 165, 812–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, I.M.; Koricanac, L.B.; Todorovic, D.V.; Ristic-Fira, A.M.; Valastro, L.M. Privitera, G. Cuttone, G. Viability of a human melanoma cell after single and combined treatment with fotemustine, dacarbazine, and proton irradiation. Ann. N. Y. Acad. Sci. 2007, 1095, 154–164. [Google Scholar] [CrossRef]
- Giallongo, A.; Oliva, D.; Cali, L.; Barba, G.; Barbieri, G.; Feo, S. Structure of the human gene for alpha-enolase. Eur. J. Biochem. 1990, 190, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Oliva, D.; Cali, L.; Feo, S.; Giallongo, A. Complete structure of the human gene encoding neuron-specific enolase. Genomics 1991, 10, 157–165. [Google Scholar] [CrossRef]
- Song, Y.; Luo, Q.; Long, H.; Hu, Z.; Que, T.; Zhang, X.; Li, Z.; Wang, G.; Yi, L.; Liu, Z.; et al. Alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol. Cancer 2015, 13, 235. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.; Kim, H.; Lee, N.; Won, C.; Kim, H.R.; Hwang, Y.I.; Song, Y.W.; Kang, J.S.; Lee, W.J. alpha-Enolase expressed on the surfaces of monocytes and macrophages induces robust synovial inflammation in rheumatoid arthritis. J. Immunol. 2012, 189, 365–372. [Google Scholar] [CrossRef]
- Choi, J.; Kim, H.; Kim, Y.; Jang, M.; Jeon, J.; Hwang, Y.I.; Shon, W.J.; Song, Y.W.; Kang, J.S.; Lee, W.J. The Anti-inflammatory Effect of GV1001 Mediated by the Downregulation of ENO1-induced Pro-inflammatory Cytokine Production. Immune Netw. 2015, 15, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Kim, J.H.; Sun, B.K.; Song, S.U.; Suh, W.; Sung, J.H. Hypoxia induces glucose uptake and metabolism of adipose derived stem cells. Mol. Med. Rep. 2016, 14, 4706–4714. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.H.; Chang, C.C.; Chen, C.S.; Tam, K.W.; Wang, Y.J.; Lee, C.H.; Lin, H.W.; Cheng, T.C.; Huang, C.S.; Chu, J.S.; et al. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 539–553. [Google Scholar] [CrossRef]
- Chang, G.C.; Liu, K.J.; Hsieh, C.L.; Hu, T.S.; Charoenfuprasert, S.; Liu, H.K.; Luh, K.T.; Hsu, L.H.; Wu, C.W.; Ting, C.C.; et al. Identification of alpha-enolase as an autoantigen in lung cancer: its overexpression is associated with clinical outcomes. Clin. Cancer Res. 2006, 12, 5746–5754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, I.; Azzouzi, A.R.; Catto, J.W.; Allen, S.; Cross, S.S.; Feeley, K.; Meuth, M.; Hamdy, F.C. Proteomic analysis of voided urine after prostatic massage from patients with prostate cancer: A pilot study. Urology 2004, 64, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Shi, J.; Cheng, S.; Zhu, Y.; Zhao, X.; Yang, K.; Du, X.; Klocker, H.; Yang, X.; Zhang, J. Estrogen promotes prostate cancer cell migration via paracrine release of ENO1 from stromal cells. Mol. Endocrinol. 2012, 26, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramani, M.; Nakao, C.; Uechi, G.T.; Cardamone, J.; Kamath, K.; Leslie, K.L.; Balachandran, R.; Wilson, L.; Day, B.W.; Jordan, M.A. Characterization and detection of cellular and proteomic alterations in stable stathmin-overexpressing, taxol-resistant BT549 breast cancer cells using offgel IEF/PAGE difference gel electrophoresis. Mutation Res. 2011, 722, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Konieczna, A.; Szczepanska, A.; Sawiuk, K.; Wegrzyn, G.; Lyzen, R. Effects of partial silencing of genes coding for enzymes involved in glycolysis and tricarboxylic acid cycle on the enterance of human fibroblasts to the S phase. BMC Cell Biol. 2015, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Feo, S.; Arcuri, D.; Piddini, E.; Passantino, R.; Giallongo, A. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 2000, 473, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, L.; Endoh, A.; Hummelke, G.; Hawks, C.L.; Hornsby, P.J. Identification of alpha-enolase as a nuclear DNA-binding protein in the zona fasciculata but not the zona reticularis of the human adrenal cortex. J. Endocrinol. 2005, 184, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, T.; Iizuka, N.; Tsunedomi, R.; Hamamoto, Y.; Miyamoto, T.; Iida, M.; Tokuhisa, Y.; Sakamoto, K.; Takashima, M.; Tamesa, T.; et al. Glycolysis module activated by hypoxia-inducible factor 1alpha is related to the aggressive phenotype of hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 725–731. [Google Scholar]
- Schreier, B.; Hocker, B. Engineering the enolase magnesium II binding site: implications for its evolution. Biochemistry 2010, 49, 7582–7589. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lehar, J.; Stockwell, B.R.; Giaever, G.; Nislow, C. Combination chemical genetics. Nat. Chem. Biol. 2008, 4, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisy, A.A.; Elliott, P.J.; Hurst, N.W.; Lee, M.S.; Lehar, J.; Price, E.R.; Serbedzija, G.; Zimmermann, G.R.; Foley, M.A.; Stockwell, B.R.; et al. Systematic discovery of multicomponent therapeutics. Proc. Natl. Acad. Sci. USA 2003, 100, 7977–7982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brough, R.; Frankum, J.R.; Costa-Cabral, S.; Lord, C.J.; Ashworth, A. Searching for synthetic lethality in cancer. Curr. Opin. Genet. Dev. 2011, 21, 34–41. [Google Scholar] [CrossRef]
- Fang, R.; Cui, Q.; Sun, J.; Duan, X.; Ma, X.; Wang, W.; Cheng, B.; Liu, Y.; Hou, Y.; Bai, G. PDK1/Akt/PDE4D axis identified as a target for asthma remedy synergistic with beta2 AR agonists by a natural agent arctigenin. Allergy 2015, 70, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Gao, J.; Cheng, C.; Zhang, M.; Liu, W.; Ma, X.; Lei, W.; Hao, E.; Hou, X.; Hou, Y.; et al. Cinnamaldehyde Enhances Antimelanoma Activity through Covalently Binding ENO1 and Exhibits a Promoting Effect with Dacarbazine. Cancers 2020, 12, 311. https://doi.org/10.3390/cancers12020311
Zhang W, Gao J, Cheng C, Zhang M, Liu W, Ma X, Lei W, Hao E, Hou X, Hou Y, et al. Cinnamaldehyde Enhances Antimelanoma Activity through Covalently Binding ENO1 and Exhibits a Promoting Effect with Dacarbazine. Cancers. 2020; 12(2):311. https://doi.org/10.3390/cancers12020311
Chicago/Turabian StyleZhang, Weiyi, Jie Gao, Chuanjing Cheng, Man Zhang, Wenjuan Liu, Xiaoyao Ma, Wei Lei, Erwei Hao, Xiaotao Hou, Yuanyuan Hou, and et al. 2020. "Cinnamaldehyde Enhances Antimelanoma Activity through Covalently Binding ENO1 and Exhibits a Promoting Effect with Dacarbazine" Cancers 12, no. 2: 311. https://doi.org/10.3390/cancers12020311
APA StyleZhang, W., Gao, J., Cheng, C., Zhang, M., Liu, W., Ma, X., Lei, W., Hao, E., Hou, X., Hou, Y., & Bai, G. (2020). Cinnamaldehyde Enhances Antimelanoma Activity through Covalently Binding ENO1 and Exhibits a Promoting Effect with Dacarbazine. Cancers, 12(2), 311. https://doi.org/10.3390/cancers12020311