Selective Killing of Activated T Cells by 5-Aminolevulinic Acid Mediated Photodynamic Effect: Potential Improvement of Extracorporeal Photopheresis

 , ,
, , 
Abstract
:
1. Introduction
2. Results
2.1. Cell Cycle, Mitosis, Cell Proliferation, and ALA-Induced PpIX Production after T Cell Activation
2.2. Effects of the Parameters on ALA-Induced PpIX Production
2.3. ALA-PpIX-Mediated Apoptosis and Necrosis
2.4. Comparison between ALA/Blue Light and 8-MOP/UV-A
2.5. ALA-PpIX Production and PDT of Subpopulations of T Cells
2.6. ALA-PDT of Mixed Populations of Resting and Activated T Cells
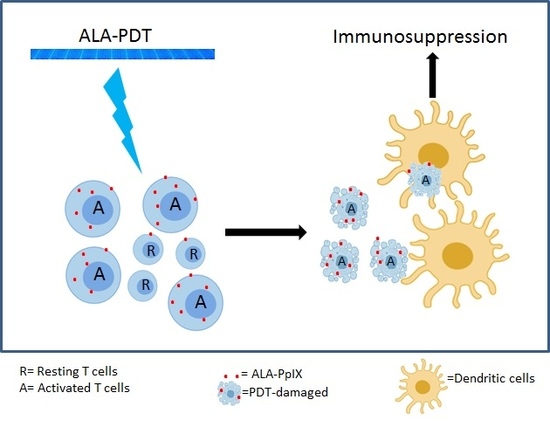
2.7. Monocyte-Derived DCs Are Resistant to ALA-PDT
2.8. ALA-PDT-Treated T Cells Induce Tolerogenic DCs
2.9. ALA-PDT-Treated T Cells Induce the Expression of Indoleamine 2, 3-Dioxygenase
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Isolation of PBMCs
4.3. Activation of T-Cells In Vitro
4.4. Fluorescence Microscopy
4.5. Flow Cytometry Analyses
4.6. CyTOF
4.7. Intracellular ALA-Induced PpIX
4.8. Light Sources
4.9. PDT Treatment Ex Vivo
4.10. CellTiter Glo® (CTG) Luminescent Cell Viability Assay
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Western Blots
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison between Cytof and Flow Data | |||
|---|---|---|---|
| Cell types | Marks | Cytof | Flow |
| Resting | CD3 | 82.3% | 88.8% |
| Resting + ALA | 84.6% | 88.4% | |
| Activated | 94.3% | 94.7% | |
| Activated + ALA | 94.2% | 96.3% | |
| Resting | CD4 | 54.0% | 57.1% |
| Resting + ALA | 56.6% | 57.6% | |
| Activated | 64.8% | 58.7% | |
| Activated + ALA | 65.2% | 58.6% | |
| Resting | CD8 | 47.5% | 31.0% |
| Resting + ALA | 46.8% | 30.6% | |
| Activated | 41.1% | 33.8% | |
| Activated + ALA | 40.8% | 33.8% | |
| Resting | CD25 | 2.18% | 3.00% |
| Resting + ALA | 2.64% | 3.08% | |
| Activated | 97.3% | 96.9% | |
| Activated + ALA | 97.9% | 97.2% | |
References
- Edelson, R.; Berger, C.; Gasparro, F.; Jegasothy, B.; Heald, P.; Wintroub, B.; Vonderheid, E.; Knobler, R.; Wolff, K.; Plewig, G.; et al. Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. Preliminary results. New Engl. J. Med. 1987, 316, 297–303. [Google Scholar] [CrossRef]
- Alfred, A.; Taylor, P.C.; Dignan, F.; El-Ghariani, K.; Griffin, J.; Gennery, A.R.; Bonney, D.; Das-Gupta, E.; Lawson, S.; Malladi, R.K.; et al. The role of extracorporeal photopheresis in the management of cutaneous T-cell lymphoma, graft-versus-host disease and organ transplant rejection: A consensus statement update from the UK Photopheresis Society. Br. J. Haematol. 2017, 177, 287–310. [Google Scholar] [CrossRef] [Green Version]
- Trautinger, F.; Just, U.; Knobler, R. Photopheresis (extracorporeal photochemotherapy). Photochem. Photobiol. Sci.: Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2013, 12, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M. Extracorporeal photopheresis: How, when, and why. J. Clin. Apher. 2011, 26, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, E.; Varela, I.; Tsirigotis, P. Update on the mechanism of action and on clinical efficacy of extracorporeal photopheresis in the treatment of acute and chronic graft versus host disease in children. Transfus. Apher. Sci.: Off. J. World Apher. Assoc.: Off. J. Eur. Soc. Haemapheresis 2012, 46, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, T.A.; Stuart, P.M.; Herndon, J.M.; Griffith, T.S. Apoptosis, tolerance, and regulatory T cells--old wine, new wineskins. Immunol. Rev. 2003, 193, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Fehervari, Z.; Sakaguchi, S. Development and function of CD25+CD4+ regulatory T cells. Curr. Opin. Immunol. 2004, 16, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Di Biaso, I.; Leoni, V.; Gaipa, G.; Rossi, V.; Bugarin, C.; Renoldi, G.; Parma, M.; Balduzzi, A.; Perseghin, P.; et al. Extracorporeal photochemotherapy is accompanied by increasing levels of circulating CD4+CD25+GITR+Foxp3+CD62L+ functional regulatory T-cells in patients with graft-versus-host disease. Transplantation 2007, 84, 31–39. [Google Scholar] [CrossRef]
- Wolff, K. Side-effects of psoralen photochemotherapy (PUVA). Br. J. Dermatol. 1990, 122 (Suppl. 36), 117–125. [Google Scholar] [CrossRef]
- Archier, E.; Devaux, S.; Castela, E.; Gallini, A.; Aubin, F.; Le Maitre, M.; Aractingi, S.; Bachelez, H.; Cribier, B.; Joly, P.; et al. Carcinogenic risks of psoralen UV-A therapy and narrowband UV-B therapy in chronic plaque psoriasis: A systematic literature review. J. Eur. Acad. Dermatol. Venereol.: JEADV 2012, 26, 22–31. [Google Scholar] [CrossRef]
- Stern, R.S.; Thibodeau, L.A.; Kleinerman, R.A.; Parrish, J.A.; Fitzpatrick, T.B. Risk of cutaneous carcinoma in patients treated with oral methoxsalen photochemotherapy for psoriasis. New Engl. J. Med. 1979, 300, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.S.; Lange, R. Non-melanoma skin cancer occurring in patients treated with PUVA five to ten years after first treatment. J. Investig. Dermatol. 1988, 91, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostinis, P.; Vantieghem, A.; Merlevede, W.; de Witte, P.A. Hypericin in cancer treatment: More light on the way. Int. J. Biochem. Cell Biol. 2002, 34, 221–241. [Google Scholar] [CrossRef]
- Agostinis, P.; Buytaert, E.; Breyssens, H.; Hendrickx, N. Regulatory pathways in photodynamic therapy induced apoptosis. Photochem. Photobiol. Sci.: Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2004, 3, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Berg, K.; Moan, J.; Kongshaug, M.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy: Principles and experimental research. Photochem. Photobiol. 1997, 65, 235–251. [Google Scholar] [CrossRef]
- Peng, Q.; Warloe, T.; Berg, K.; Moan, J.; Kongshaug, M.; Giercksky, K.E.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy: Clinical research and future challenges. Cancer: Interdiscip. Int. J. Am. Cancer Soc. 1997, 79, 2282–2308. [Google Scholar] [CrossRef]
- Holien, T.; Gederaas, O.A.; Darvekar, S.R.; Christensen, E.; Peng, Q. Comparison between 8-methoxypsoralen and 5-aminolevulinic acid in killing T cells of photopheresis patients ex vivo. Lasers Surg. Med. 2018, 50, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Hryhorenko, E.A.; Rittenhouse-Diakun, K.; Harvey, N.S.; Morgan, J.; Stewart, C.C.; Oseroff, A.R. Characterization of endogenous protoporphyrin IX induced by delta-aminolevulinic acid in resting and activated peripheral blood lymphocytes by four-color flow cytometry. Photochem. Photobiol. 1998, 67, 565–572. [Google Scholar] [CrossRef]
- Malik, Z.; Lugaci, H. Destruction of erythroleukaemic cells by photoactivation of endogenous porphyrins. Br. J. cancer 1987, 56, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Morton, C.A.; Szeimies, R.M.; Sidoroff, A.; Braathen, L.R. European guidelines for topical photodynamic therapy part 1: Treatment delivery and current indications - actinic keratoses, Bowen’s disease, basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol.: JEADV 2013, 27, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; Group, A.L.-G.S. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncology 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Braun, D.; Longman, R.S.; Albert, M.L. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood 2005, 106, 2375–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nature Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Furset, G.; Floisand, Y.; Sioud, M. Impaired expression of indoleamine 2, 3-dioxygenase in monocyte-derived dendritic cells in response to Toll-like receptor-7/8 ligands. Immunology 2008, 123, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Rittenhouse-Diakun, K.; Van Leengoed, H.; Morgan, J.; Hryhorenko, E.; Paszkiewicz, G.; Whitaker, J.E.; Oseroff, A.R. The role of transferrin receptor (CD71) in photodynamic therapy of activated and malignant lymphocytes using the heme precursor delta-aminolevulinic acid (ALA). Photochem. Photobiol. 1995, 61, 523–528. [Google Scholar] [CrossRef]
- Furre, I.E.; Shahzidi, S.; Luksiene, Z.; Moller, M.T.; Borgen, E.; Morgan, J.; Tkacz-Stachowska, K.; Nesland, J.M.; Peng, Q. Targeting PBR by hexaminolevulinate-mediated photodynamic therapy induces apoptosis through translocation of apoptosis-inducing factor in human leukemia cells. Cancer Res. 2005, 65, 11051–11060. [Google Scholar] [CrossRef] [Green Version]
- Morelli, A.E.; Larregina, A.T. Concise Review: Mechanisms Behind Apoptotic Cell-Based Therapies Against Transplant Rejection and Graft versus Host Disease. Stem Cells 2016, 34, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [Green Version]
- Futterleib, J.S.; Feng, H.; Tigelaar, R.E.; Choi, J.; Edelson, R.L. Activation of GILZ gene by photoactivated 8-methoxypsoralen: Potential role of immunoregulatory dendritic cells in extracorporeal photochemotherapy. Transfus. Apher. Sci.: Off. J. World Apher. Assoc.: Off. J. Eur. Soc. Haemapheresis 2014, 50, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Trickett, A.; Kwan, Y.L. T cell stimulation and expansion using anti-CD3/CD28 beads. J. Immunol. Methods 2003, 275, 251–255. [Google Scholar] [CrossRef]
- Cunderlikova, B.; Vasovic, V.; Randeberg, L.L.; Christensen, E.; Warloe, T.; Nesland, J.M.; Peng, Q. Modification of extracorporeal photopheresis technology with porphyrin precursors. Comparison between 8-methoxypsoralen and hexaminolevulinate in killing human T-cell lymphoma cell lines in vitro. Biochim. Biophys. Acta 2014, 1840, 2702–2708. [Google Scholar] [CrossRef] [PubMed]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darvekar, S.; Juzenas, P.; Oksvold, M.; Kleinauskas, A.; Holien, T.; Christensen, E.; Stokke, T.; Sioud, M.; Peng, Q. Selective Killing of Activated T Cells by 5-Aminolevulinic Acid Mediated Photodynamic Effect: Potential Improvement of Extracorporeal Photopheresis. Cancers 2020, 12, 377. https://doi.org/10.3390/cancers12020377
Darvekar S, Juzenas P, Oksvold M, Kleinauskas A, Holien T, Christensen E, Stokke T, Sioud M, Peng Q. Selective Killing of Activated T Cells by 5-Aminolevulinic Acid Mediated Photodynamic Effect: Potential Improvement of Extracorporeal Photopheresis. Cancers. 2020; 12(2):377. https://doi.org/10.3390/cancers12020377
Chicago/Turabian StyleDarvekar, Sagar, Petras Juzenas, Morten Oksvold, Andrius Kleinauskas, Toril Holien, Eidi Christensen, Trond Stokke, Mouldy Sioud, and Qian Peng. 2020. "Selective Killing of Activated T Cells by 5-Aminolevulinic Acid Mediated Photodynamic Effect: Potential Improvement of Extracorporeal Photopheresis" Cancers 12, no. 2: 377. https://doi.org/10.3390/cancers12020377
APA StyleDarvekar, S., Juzenas, P., Oksvold, M., Kleinauskas, A., Holien, T., Christensen, E., Stokke, T., Sioud, M., & Peng, Q. (2020). Selective Killing of Activated T Cells by 5-Aminolevulinic Acid Mediated Photodynamic Effect: Potential Improvement of Extracorporeal Photopheresis. Cancers, 12(2), 377. https://doi.org/10.3390/cancers12020377