Physiological and Pathological Roles of RAD52 at DNA Replication Forks


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
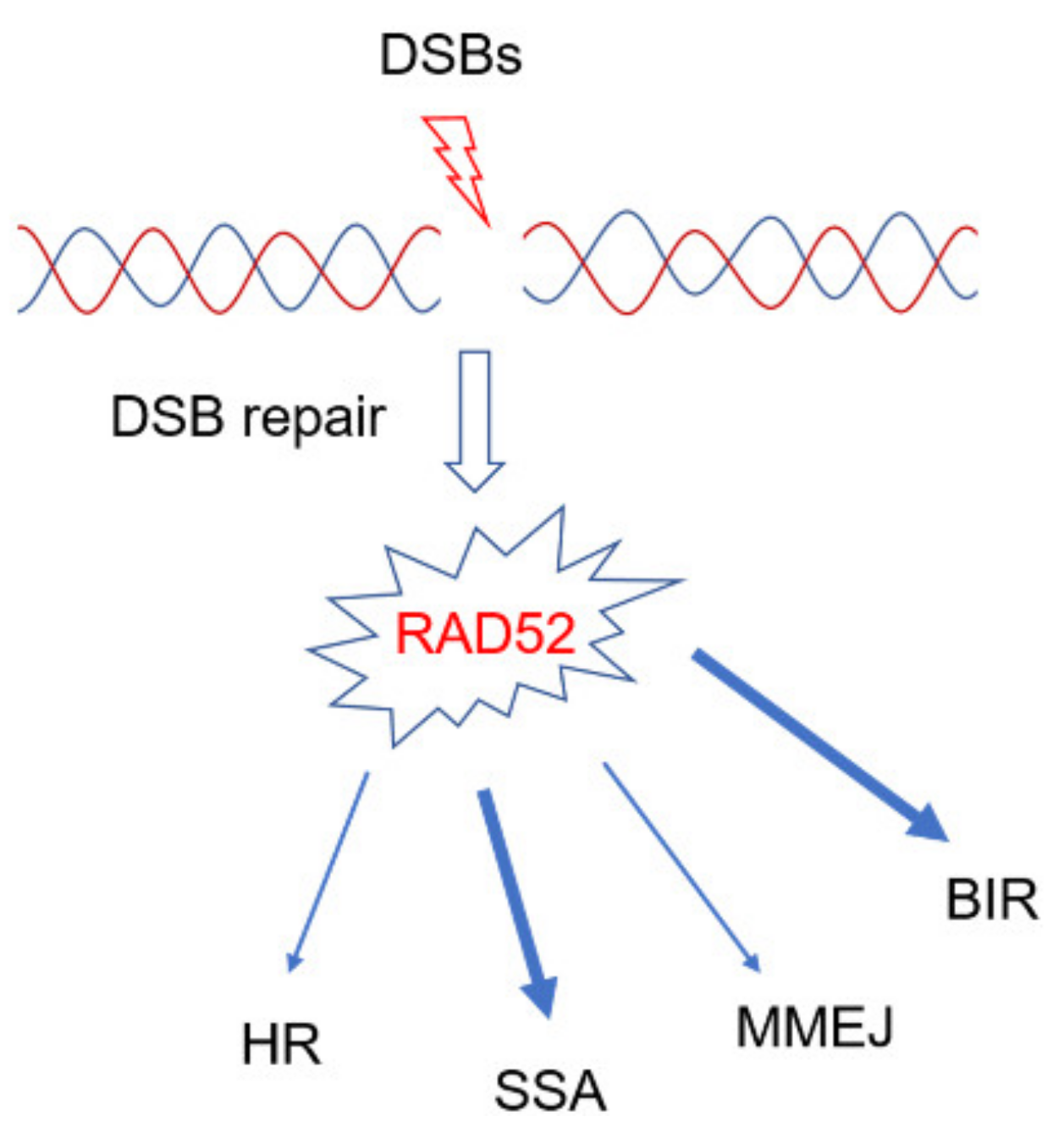
2. Role of RAD52 in DSBs Repair
3. Regulation of RAD52 and its Relevance in DSB Repair
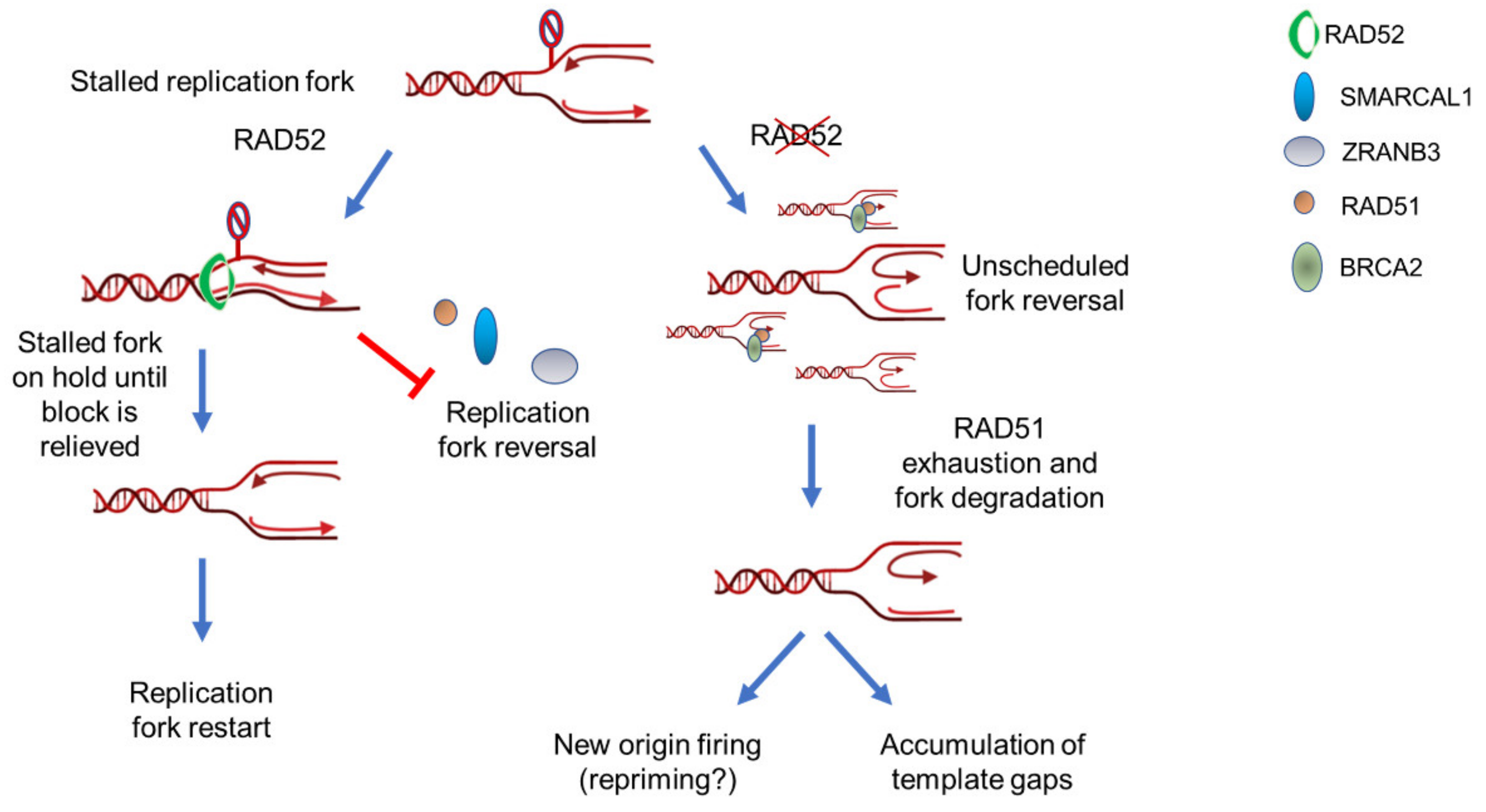
4. Replication Fork Recovery
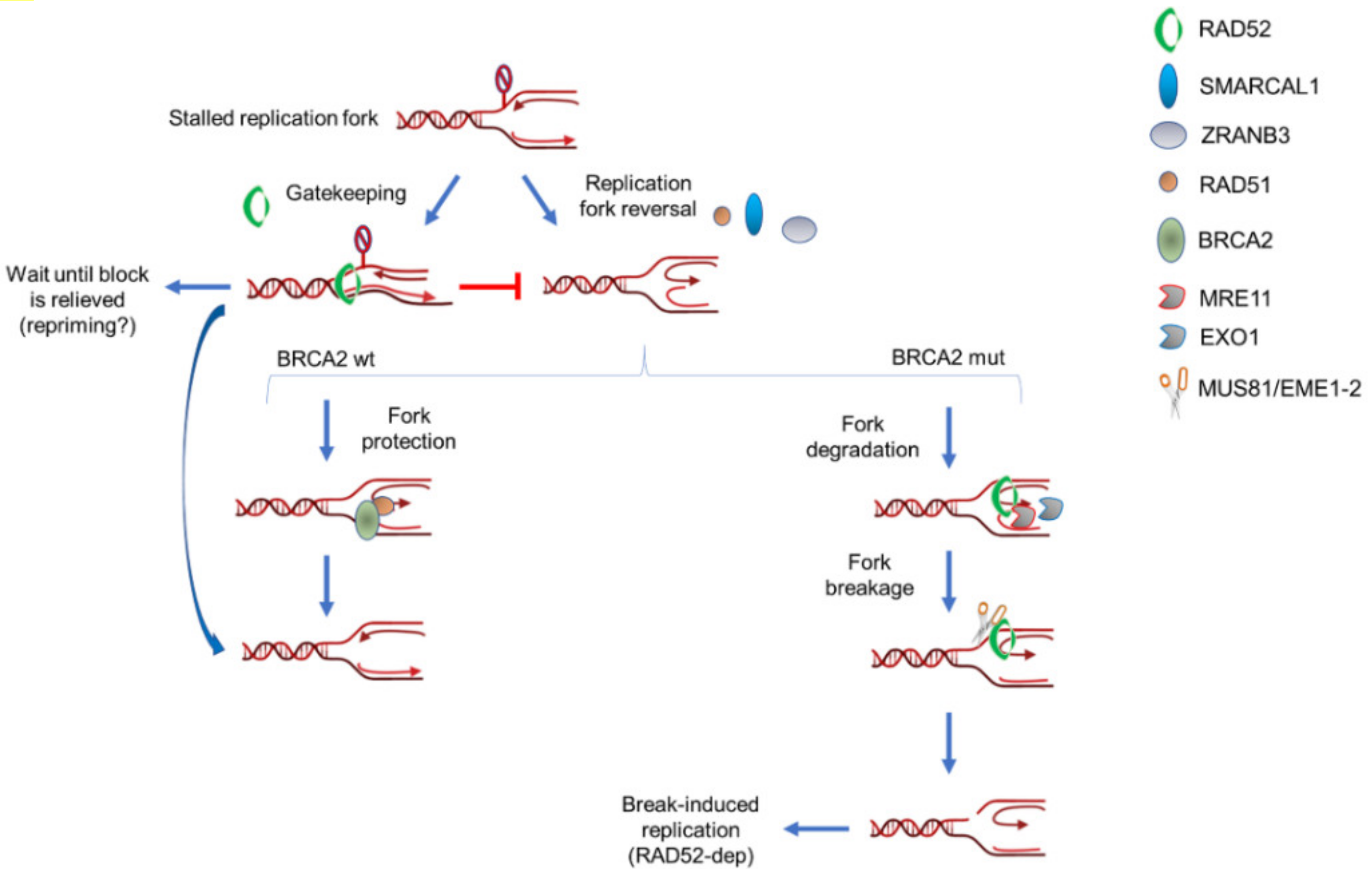
5. RAD52 as “Gatekeeper“ of Perturbed Replication Fork
6. RAD52 as a Partner of MUS81
7. Conclusions and Potential Implications of Replication Fork-Related RAD52 Roles in Cancer Therapy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchitto, A.; Pichierri, P. Understanding the molecular basis of common fragile sites instability: Role of the proteins involved in the recovery of stalled replication forks. Cell Cycle 2011, 10, 4039–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, e22. [Google Scholar] [CrossRef] [Green Version]
- Franchitto, A.; Pichierri, P. Replication fork recovery and regulation of common fragile sites stability. Cell. Mol. Life Sci. 2014, 71, 4507–4517. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazina, O.; Mazin, A. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes (Basel) 2016, 7, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalan, M.; Olsen, K.S.; Powell, S.N. Emerging Roles of RAD52 in Genome Maintenance. Cancers (Basel) 2019, 11, e1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003, 4, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuzuki, T.; Fujii, Y.; Sakumi, K.; Tominaga, Y.; Nakao, K.; Sekiguchi, M.; Matsushiro, A.; Yoshimura, Y.; Morita, T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6236–6240. [Google Scholar] [CrossRef] [Green Version]
- Rijkers, T.; Van Den Ouweland, J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.; Lohman, P.H.; Pastink, A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Buerstedde, J.M.; Bezzubova, O.; Morrison, C.; Takata, M.; Shinohara, A.; Takeda, S. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 1998, 18, 6430–6435. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, L.; Venkitaraman, A. Emerging functions of BRCA2 in DNA recombination. Trends Biochem. Sci. 2004, 29, 310–316. [Google Scholar] [CrossRef]
- Dray, E.; Siaud, N.; Dubois, E.; Doutriaux, M.-P. Interaction between Arabidopsis Brca2 and its partners Rad51, Dmc1, and Dss1. Plant Physiol. 2006, 140, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Kojic, M.; Kostrub, C.F.; Buchman, A.R.; Holloman, W.K. BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis. Mol. Cell 2002, 10, 683–691. [Google Scholar] [CrossRef]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol. Cell. Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2012, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutt, A.; Ashworth, A. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol. Med. 2002, 8, 571–576. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Wu, X. Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers (Basel) 2019, 11, e1467. [Google Scholar] [CrossRef] [Green Version]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers (Basel) 2019, 11, e1561. [Google Scholar] [CrossRef] [Green Version]
- Franchet, C.; Hoffmann, J.-S. When RAD52 Allows Mitosis to Accept Unscheduled DNA Synthesis. Cancers (Basel) 2019, 12, e26. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability — an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2015, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Kwon, Y.H.; Niu, H.; Sung, P. Investigations of homologous recombination pathways and their regulation. Yale J. Biol. Med. 2013, 86, 453–461. [Google Scholar] [PubMed]
- Wray, J.; Liu, J.; Nickoloff, J.A.; Shen, Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res. 2008, 68, 2699–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Qing, Y.; Yamazoe, M.; Hirota, K.; Dejsuphong, D.; Sakai, W.; Yamamoto, K.N.; Bishop, D.K.; Wu, X.; Takeda, S. The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet. 2011, 7, e1002148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Scully, R.; Ganesan, S.; Vlasakova, K.; Chen, J.; Socolovsky, M.; Livingston, D.M. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell 1999, 4, 1093–1099. [Google Scholar] [CrossRef]
- Grimme, J.M.; Honda, M.; Wright, R.; Okuno, Y.; Rothenberg, E.; Mazin, A.V.; Ha, T.; Spies, M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010, 38, 2917–2930. [Google Scholar] [CrossRef] [Green Version]
- Pokhrel, N.; Caldwell, C.C.; Corless, E.I.; Tillison, E.A.; Tibbs, J.; Jocic, N.; Tabei, S.M.A.; Wold, M.S.; Spies, M.; Antony, E. Dynamics and selective remodeling of the DNA-binding domains of RPA. Nat. Struct. Mol. Biol. 2019, 26, 129–136. [Google Scholar] [CrossRef]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 0, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signon, L.; Malkova, A.; Naylor, M.L.; Klein, H.; Haber, J.E. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell. Biol. 2001, 21, 2048–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Ré, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, e860. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Claussin, C.; Chang, M. Multiple Rad52-Mediated Homology-Directed Repair Mechanisms Are Required to Prevent Telomere Attrition-Induced Senescence in Saccharomyces cerevisiae. PLoS Genet. 2016, 12, e1006176. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanyam, S.; Ismail, M.; Bhattacharya, I.; Spies, M. Tyrosine phosphorylation stimulates activity of human RAD51 recombinase through altered nucleoprotein filament dynamics. Proc. Natl. Acad. Sci. 2016, 113, e201604807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Yuan, S.S.; Liu, W.; Xu, Y.; Trujillo, K.; Song, B.; Cong, F.; Goff, S.P.; Wu, Y.; Arlinghaus, R.; et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 1999, 274, 12748–12752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitao, H.; Yuan, Z.-M. Regulation of ionizing radiation-induced Rad52 nuclear foci formation by c-Abl-mediated phosphorylation. J. Biol. Chem. 2002, 277, 48944–48948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskaran, R.; Wood, L.D.; Whitaker, L.L.; Canman, C.E.; Morgan, S.E.; Xu, Y.; Barlow, C.; Baltimore, D.; Wynshaw-Boris, A.; Kastan, M.B.; et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997, 387, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.S.; Plattner, R. Activation of abl family kinases in solid tumors. Genes Cancer 2012, 3, 414–425. [Google Scholar] [CrossRef]
- Altmannova, V.; Eckert-Boulet, N.; Arneric, M.; Kolesar, P.; Chaloupkova, R.; Damborsky, J.; Sung, P.; Zhao, X.; Lisby, M.; Krejci, L. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 2010, 38, 4708–4721. [Google Scholar] [CrossRef] [Green Version]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar] [CrossRef]
- Stefanovie, B.; Hengel, S.R.; Mlcouskova, J.; Prochazkova, J.; Spirek, M.; Nikulenkov, F.; Nemecek, D.; Koch, B.G.; Bain, F.E.; Yu, L.; et al. DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic Acids Res. 2019, 1–15. [Google Scholar] [CrossRef]
- Barroso-González, J.; García-Expósito, L.; Hoang, S.M.; Lynskey, M.L.; Roncaioli, J.L.; Ghosh, A.; Wallace, C.T.; Modesti, M.; Bernstein, K.A.; Sarkar, S.N.; et al. RAD51AP1 Is an Essential Mediator of Alternative Lengthening of Telomeres. Mol. Cell 2019, 76, 11–26. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [Green Version]
- Unk, I.; Hajdú, I.; Blastyák, A.; Haracska, L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst). 2010, 9, 257–267. [Google Scholar] [CrossRef]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.-W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Achar, Y.J.; Balogh, D.; Haracska, L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA 2011, 108, 14073–14078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, L.A.; Cortez, D. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Groden, J.; Orren, D.K. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 2006, 45, 13939–13946. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Décaillet, C.; Delannoy, M.; Wu, L.; Constantinou, A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. USA 2008, 105, 16107–16112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bétous, R.; Couch, F.; Mason, A.; Eichman, B.; Manosas, M.; Cortez, D. Substrate-Selective Repair and Restart of Replication Forks by DNA Translocases. Cell Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansbach, C.E.; Bétous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; de Antoni, A.; Técher, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, e859. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [Green Version]
- Hengel, S.R.; Malacaria, E.; Folly da Silva Constantino, L.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife 2016, 5, e14740. [Google Scholar] [CrossRef]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef] [Green Version]
- Bétous, R.; Goullet de Rugy, T.; Pelegrini, A.L.; Queille, S.; de Villartay, J.-P.; Hoffmann, J.-S. DNA replication stress triggers rapid DNA replication fork breakage by Artemis and XPF. PLoS Genet. 2018, 14, e1007541. [Google Scholar] [CrossRef]
- Malacaria, E.; Franchitto, A.; Pichierri, P. SLX4 Prevents GEN1-Dependent DSBs During DNA Replication Arrest Under Pathological Conditions in Human Cells. Sci. Rep. 2017, 7, e44464. [Google Scholar] [CrossRef] [Green Version]
- Machwe, A.; Xiao, L.; Lloyd, R.G.; Bolt, E.; Orren, D.K. Replication fork regression in vitro by the Werner syndrome protein (WRN): Holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 2007, 35, 5729–5747. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Malacaria, E.; Pugliese, G.M.; Honda, M.; Marabitti, V.; Aiello, F.A.; Spies, M.; Franchitto, A.; Pichierri, P. Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 2019, 10, 1–19. [Google Scholar]
- Bhat, K.P.; Bétous, R.; Cortez, D. High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J. Biol. Chem. 2015, 290, 4110–4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagawa, W.; Kagawa, A.; Saito, K.; Ikawa, S.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Identification of a second DNA binding site in the human Rad52 protein. J. Biol. Chem. 2008, 283, 24264–24273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386. [Google Scholar] [CrossRef]
- Schubert, L.; Ho, T.; Hoffmann, S.; Haahr, P.; Guérillon, C.; Mailand, N. RADX interacts with single-stranded DNA to promote replication fork stability. EMBO Rep. 2017, 18, 1991–2003. [Google Scholar] [CrossRef]
- Pasero, P.; Vindigni, A. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Chaudhuri, A.R.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Iannascoli, C.; Palermo, V.; Murfuni, I.; Franchitto, A.; Pichierri, P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015, 43, 9788–9803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinet, A.; Tirman, S.; Jackson, J.; Šviković, S.; Lemaçon, D.; Carvajal-Maldonado, D.; González-Acosta, D.; Vessoni, A.T.; Cybulla, E.; Wood, M.; et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 2019, 77, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Vallerga, M.B.; Mansilla, S.F.; Federico, M.B.; Bertolin, A.P.; Gottifredi, V. Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E6624–E6633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murfuni, I.; Basile, G.; Subramanyam, S.; Malacaria, E.; Bignami, M.; Spies, M.; Franchitto, A.; Pichierri, P. Survival of the Replication Checkpoint Deficient Cells Requires MUS81-RAD52 Function. PLoS Genet. 2013, 9, e1003910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Pirzio, L.M.; Prosperi, E.; Sapora, O.; Bignami, M.; Pichierri, P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008, 183, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Leman, A.R.; Noguchi, C.; Lee, C.Y.; Noguchi, E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 2010, 123, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Errico, A.; Costanzo, V.; Hunt, T. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc. Natl. Acad. Sci. USA. 2007, 104, 14929–14934. [Google Scholar] [CrossRef] [Green Version]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A. Mutational signatures reveal the role of RAD52 in p53-independent p21 driven genomic instability. Genome Biol. 2018, 19, e37. [Google Scholar] [CrossRef] [Green Version]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Murfuni, I.; Nicolai, S.; Baldari, S.; Crescenzi, M.; Bignami, M.; Franchitto, A.; Pichierri, P. The WRN and MUS81 proteins limit cell death and genome instability following oncogene activation. Oncogene 2012, 32, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, C.H.; Demin, A.A.; Munashingha, P.R.; Amangyeld, T.; Kwon, B.; Formosa, T.; Seo, Y.S. Rad52/Rad59-dependent recombination as a means to rectify faulty Okazaki fragment processing. J. Biol. Chem. 2014, 289, 15064–15079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doe, C.L.; Osman, F.; Dixon, J.; Whitby, M.C. DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 2004, 32, 5570–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasparek, T.R.; Humphrey, T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011, 22, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Aguilera, A. Complex Chromosomal Rearrangements Mediated by Break-Induced Replication Involve Structure-Selective Endonucleases. PLoS Genet. 2012, 8, e1002979. [Google Scholar] [CrossRef] [Green Version]
- Vasan, S.; Deem, A.; Ramakrishnan, S.; Argueso, J.L.; Malkova, A. Cascades of Genetic Instability Resulting from Compromised Break-Induced Replication. PLoS Genet. 2014, 10, e1004119. [Google Scholar] [CrossRef] [Green Version]
- Sakofsky, C.J.; Roberts, S.A.; Malc, E.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Malkova, A. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014, 7, 1640–1648. [Google Scholar] [CrossRef] [Green Version]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Leuzzi, G.; Marabitti, V.; Pichierri, P.; Franchitto, A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016, 35, 1437–1451. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengel, S.R.; Spies, M.A.; Spies, M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem. Biol. 2017, 24, 1101–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Wakefield, M.J.; Nesic, K.; Kondrashova, O.; Scott, C.L. Diverse mechanisms of PARP inhibitor resistance in ovarian cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, e188307. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef]
- Boyer, A.-S.; Walter, D.; Sørensen, C.S. DNA replication and cancer: From dysfunctional replication origin activities to therapeutic opportunities. Semin. Cancer Biol. 2016, 37, 16–25. [Google Scholar] [CrossRef]
- Manic, G.; Sistigu, A.; Corradi, F.; Musella, M.; De Maria, R.; Vitale, I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin. Cancer Biol. 2018, 53, 31–41. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Dent, P. Investigational CHK1 inhibitors in early phase clinical trials for the treatment of cancer. Expert Opin. Investig. Drugs 2019, 28, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tomaszowski, K.-H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLθ pathways. Elife 2018, 7, e31723. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating “BRCAness”. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malacaria, E.; Honda, M.; Franchitto, A.; Spies, M.; Pichierri, P. Physiological and Pathological Roles of RAD52 at DNA Replication Forks. Cancers 2020, 12, 402. https://doi.org/10.3390/cancers12020402
Malacaria E, Honda M, Franchitto A, Spies M, Pichierri P. Physiological and Pathological Roles of RAD52 at DNA Replication Forks. Cancers. 2020; 12(2):402. https://doi.org/10.3390/cancers12020402
Chicago/Turabian StyleMalacaria, Eva, Masayoshi Honda, Annapaola Franchitto, Maria Spies, and Pietro Pichierri. 2020. "Physiological and Pathological Roles of RAD52 at DNA Replication Forks" Cancers 12, no. 2: 402. https://doi.org/10.3390/cancers12020402
APA StyleMalacaria, E., Honda, M., Franchitto, A., Spies, M., & Pichierri, P. (2020). Physiological and Pathological Roles of RAD52 at DNA Replication Forks. Cancers, 12(2), 402. https://doi.org/10.3390/cancers12020402