The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
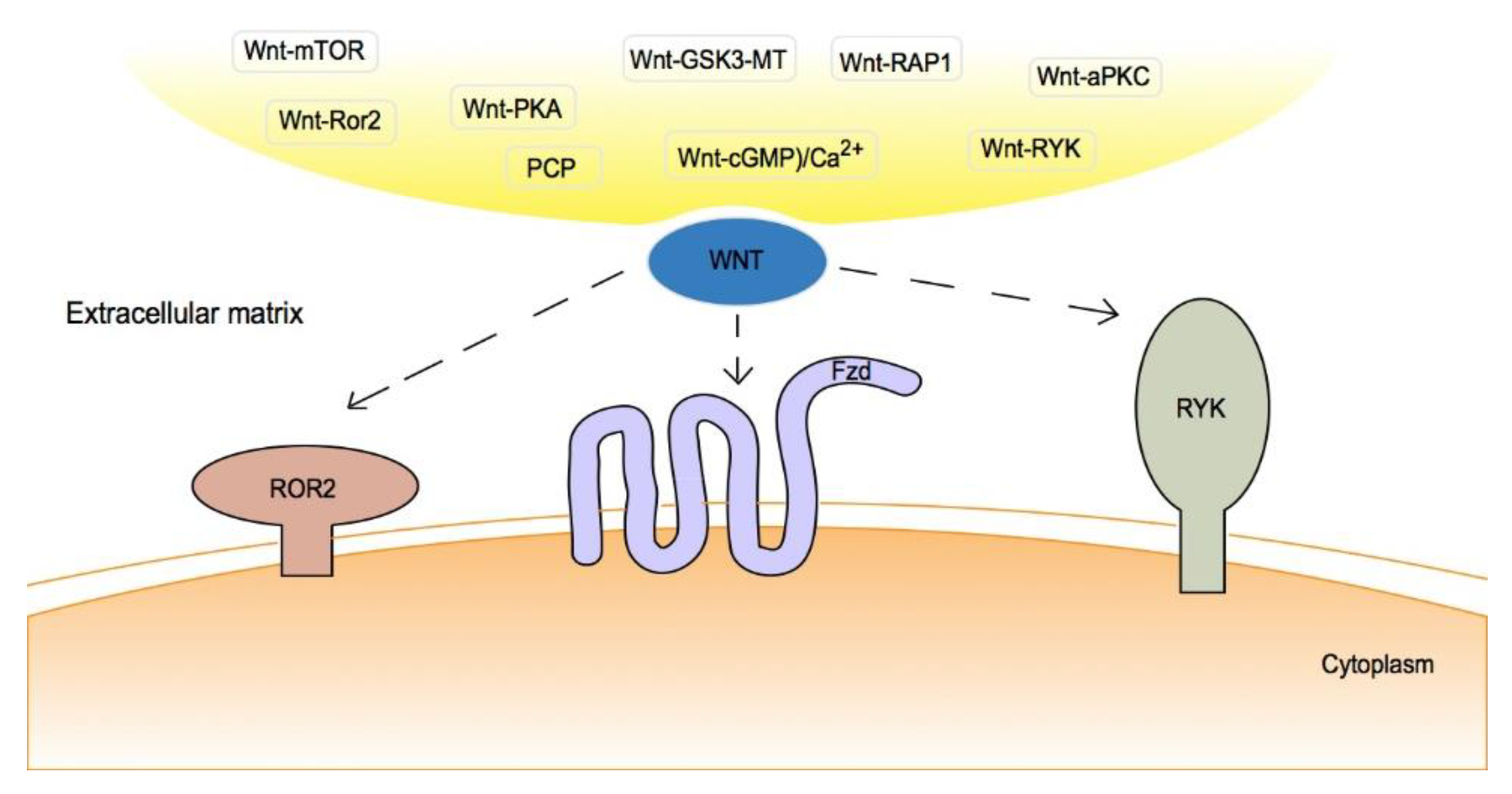
2. Canonical/Non-Canonical Wnt Signaling Pathway
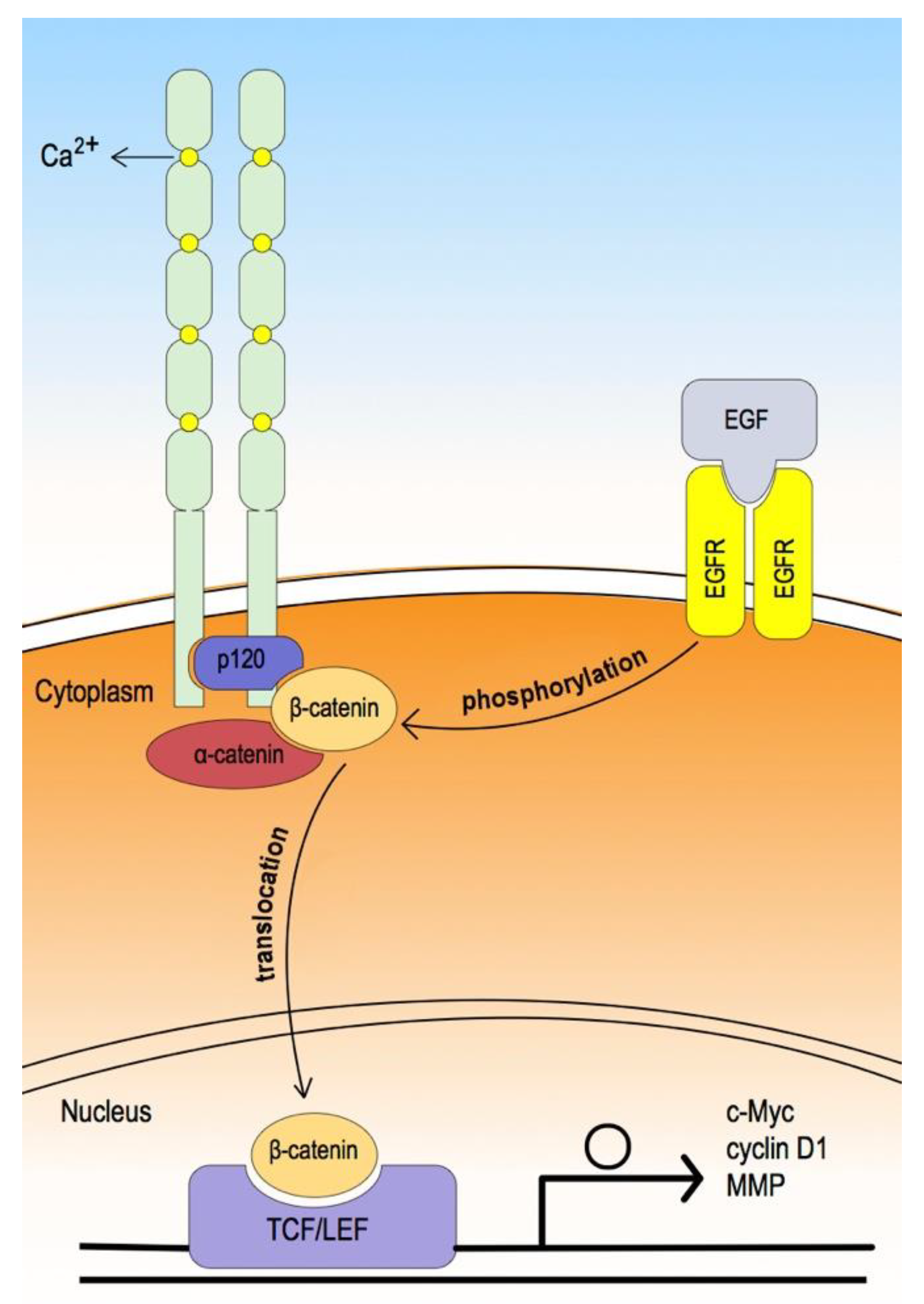
3. Epithelial Cadherin
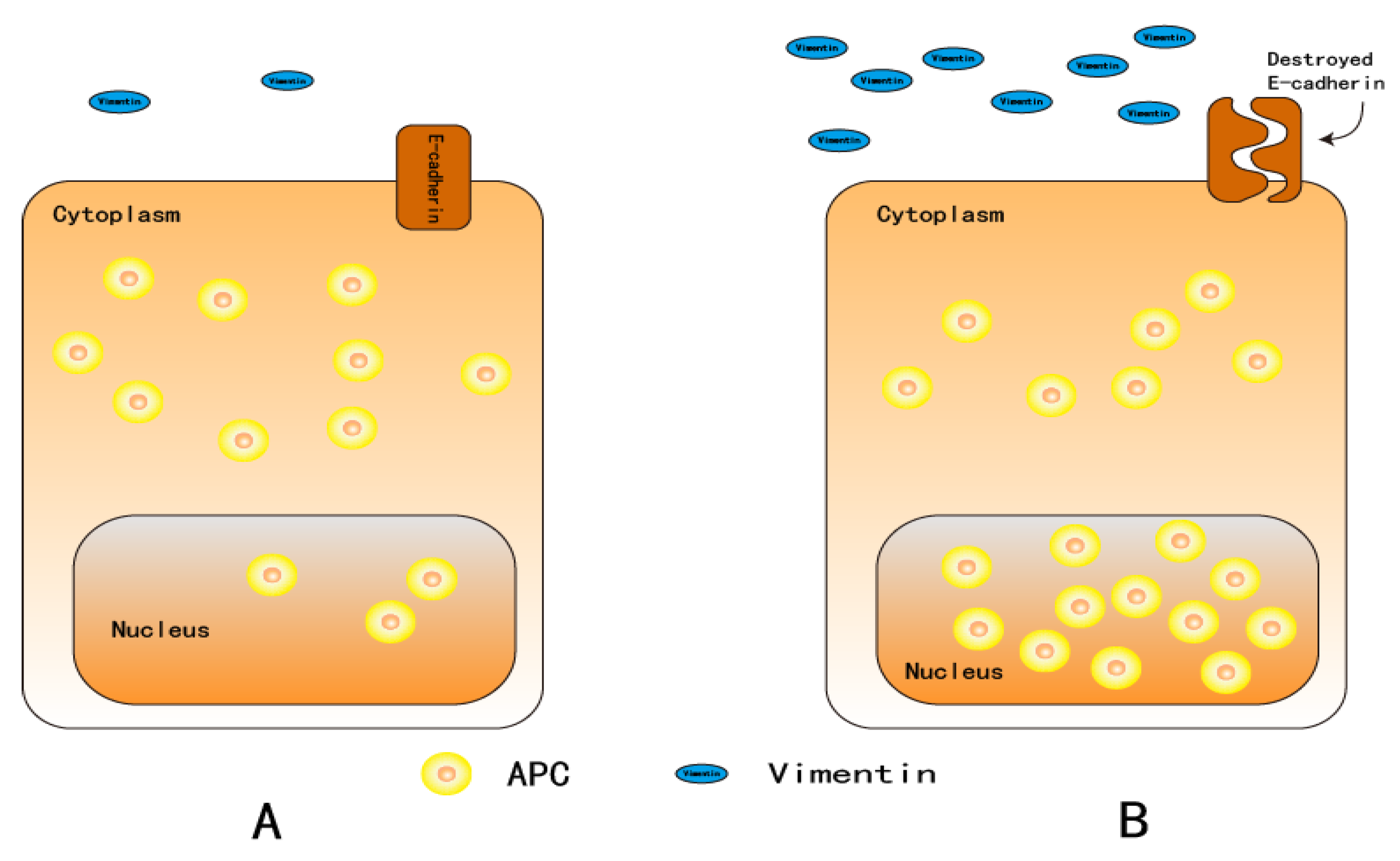
4. Vimentin
5. Adenomatous Polyposis Coli
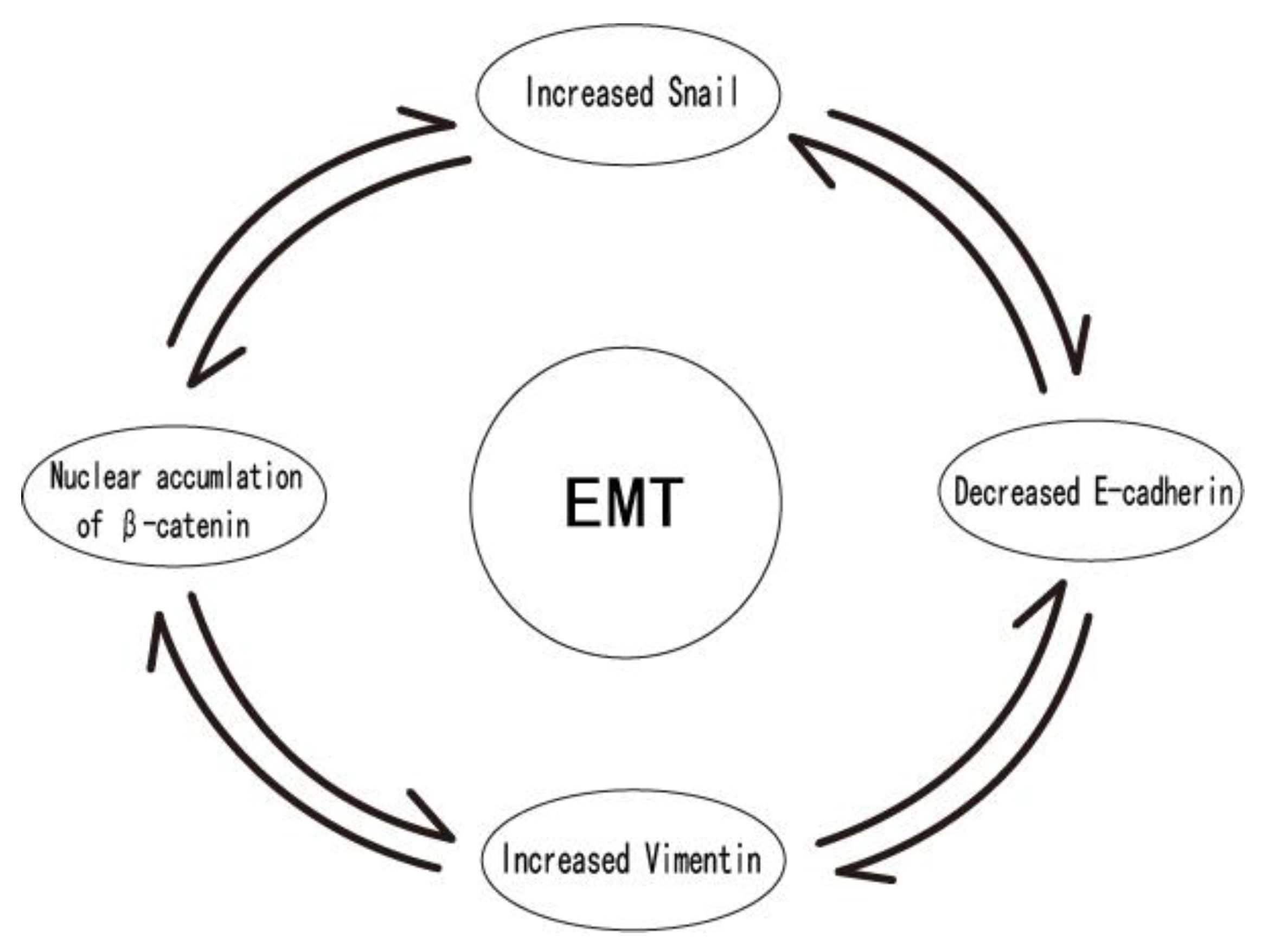
6. Snail
7. Neural Cadherin
8. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Chaw, S.Y.; Abdul Majeed, A.; Dalley, A.J.; Chan, A.; Stein, S.; Farah, C.S. Epithelial to mesenchymal transition (EMT) biomarkers—E-cadherin, beta-catenin, APC and Vimentin—In oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Glazer, C.A.; Chang, S.S.; Ha, P.K.; Califano, J.A. Applying the molecular biology and epigenetics of head and neck cancer in everyday clinical practice. Oral Oncol. 2009, 45, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Siu, A.; Lee, C.; Dang, D.; Lee, C.; Ramos, D.M. Stem cell markers as predictors of oral cancer invasion. Anticancer Res. 2012, 32, 1163–1166. [Google Scholar] [PubMed]
- Woolgar, J.A.; Triantafyllou, A. Pitfalls and procedures in the histopathological diagnosis of oral and oropharyngeal squamous cell carcinoma and a review of the role of pathology in prognosis. Oral Oncol. 2009, 45, 361–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Waal, I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009, 45, 317–323. [Google Scholar] [CrossRef]
- Van der Waal, I. Potentially malignant disorders of the oral and oropharyngeal mucosa; present concepts of management. Oral Oncol. 2010, 46, 423–425. [Google Scholar] [CrossRef]
- Mortazavi, H.; Baharvand, M.; Mehdipour, M. Oral potentially malignant disorders: An overview of more than 20 entities. J. Dent. Res. Dent. Clin. Dent. Prospects 2014, 8, 6. [Google Scholar] [CrossRef]
- Tsunematsu, K.; Nakatani, E.; Iwahashi, T.; Hideshima, K.; Karino, M.; Nariai, Y.; Kanno, T.; Kagimura, T.; Sekine, J. Feasibility of HPV16, HPV18, and p16 expression as biomarkers for distinguishing normal oral epithelium from oral epithelial dysplasia and oral intraepithelial neoplasia. Shimane J. Med. Sci. 2016, 32, 69–79. [Google Scholar]
- Sekine, J. A new conception of oral potentially malignant disorders (PMDs) from cytological and clinical viewpoints and related disorders of upper digestive tract. Shimane J. Med. Sci. 2017, 33, 39–60. [Google Scholar]
- Amagasa, T. Oral premalignant lesions. Int. J. Clin. Oncol. 2011, 16, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Johnson, N.; Van Der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa: Potentially malignant disorders. J. Oral Pathol. Med. 2007, 36, 575–580. [Google Scholar] [CrossRef]
- Casparis, S.; Borm, J.M.; Tektas, S.; Kamarachev, J.; Locher, M.C.; Damerau, G.; Grätz, K.W.; Stadlinger, B. Oral lichen planus (OLP), oral lichenoid lesions (OLL), oral dysplasia, and oral cancer: Retrospective analysis of clinicopathological data from 2002–2011. Oral Maxillofac. Surg. 2015, 19, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, R. Benign and malignant tumors of the oral cavity. In Shafer’s Textbook of Oral Pathology, 6th ed.; Elsevier: Noida, India, 2009; pp. 86–91. [Google Scholar]
- Izumo, T.; Kirita, T.; Ariji, E.; Ozeki, S.; Okada, N.; Okabe, S.; Okazaki, Y.; Omura, K.; Kusama, M.; Sato, T.; et al. General rules for clinical and pathological studies on oral cancer: A synopsis. Jpn. J. Clin. Oncol. 2012, 42, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Albers, A.E.; Qin, J.; Kaufmann, A.M. Prognostic significance of overexpressed p16INK4a in patients with cervical cancer: A meta-analysis. PLoS ONE 2014, 9, e106384. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, C.; Teleman, S.; Socolov, D.; Anton, G.; Mihailovici, M. Evaluation of p16INK4a and Ki-67 proteins expression in cervical intraepithelial neoplasia and their correlation with HPV-HR infection. Rev. Med. Chir. Soc. Med. Nat. Iasi 2010, 114, 823–828. [Google Scholar]
- Wu, J.; Li, X.-J.; Zhu, W.; Liu, X.-P. Detection and pathological value of papillomavirus DNA and p16INK4A and p53 protein expression in cervical intraepithelial neoplasia. Oncol. Lett. 2014, 7, 738–744. [Google Scholar] [CrossRef]
- González-Moles, M.A.; Ruiz-Ávila, I.; Gil-Montoya, J.A.; Plaza-Campillo, J.; Scully, C. β-Catenin in oral cancer: An update on current knowledge. Oral Oncol. 2014, 50, 818–824. [Google Scholar] [CrossRef]
- Arnés, M.; Casas Tintó, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Medrek, C.; Landberg, G.; Andersson, T.; Leandersson, K. Wnt-5a-CKIα signaling promotes β-Catenin/E-Cadherin complex formation and intercellular adhesion in human breast epithelial cells. J. Biol. Chem. 2009, 284, 10968–10979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afrem, M.C.; Margaritescu, C.; Craitoiu, M.M.; Ciuca, M.; Sarla, C.G.; Cotoi, O.S. The immunohistochemical investigations of cadherin “switch” during epithelial-mesenchymal transition of tongue squamous cell carcinoma. Rom. J. Morphol. Embryol. 2014, 55, 1049–1056. [Google Scholar]
- Prgomet, Z.; Andersson, T.; Lindberg, P. Higher expression of WNT5A protein in oral squamous cell carcinoma compared with dysplasia and oral mucosa with a normal appearance. Eur. J. Oral Sci. 2017, 125, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Van Camp, J.K.; Beckers, S.; Zegers, D.; Van Hul, W. Wnt signaling and the control of human stem cell fate. Stem Cell Rev. Rep. 2014, 10, 207–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: Implications in targeted cancer therapies. Lab. Invest. 2016, 96, 116–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, M.R.; Kugel, C.H.; Weeraratna, A.T. The Wnts of change: How Wnts regulate phenotype switching in melanoma. Biochim. Biophys. Acta BBA Rev. Cancer 2015, 1856, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumawat, K.; Gosens, R. WNT-5A: Signaling and functions in health and disease. Cell. Mol. Life Sci. 2016, 73, 567–587. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Qin, L.; Luo, Z.; Guo, Q.; Yang, L.; Liao, D. Challenging role of Wnt5a and its signaling pathway in cancer metastasis (Review). Exp. Ther. Med. 2014, 8, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-N.; Zhu, N.; Liu, C.; Wu, H.-T.; Gui, Y.; Liao, D.-F.; Qin, L. Wnt5a and its signaling pathway in angiogenesis. Clin. Chim. Acta 2017, 471, 263–269. [Google Scholar] [CrossRef]
- Schulte, G. Frizzleds and WNT/β-catenin signaling—The black box of ligand–receptor selectivity, complex stoichiometry and activation kinetics. Eur. J. Pharmacol. 2015, 763, 191–195. [Google Scholar] [CrossRef]
- Clevers, H.; Loh, K.M.; Nusse, R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Mikels, A.J.; Nusse, R. Wnts as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, T.; Nakagawa, H.; Kamberov, Y.G.; Wilder, E.L.; Klein, P.S.; Rustgi, A.K. Wnt-1 but not epidermal growth factor induces beta-catenin/T-cell factor-dependent transcription in esophageal cancer cells. Cancer Res. 2002, 62, 277–282. [Google Scholar] [PubMed]
- Bartis, D.; Csongei, V.; Weich, A.; Kiss, E.; Barko, S.; Kovacs, T.; Avdicevic, M.; D’Souza, V.K.; Rapp, J.; Kvell, K. Down-regulation of canonical and up-regulation of non-canonical Wnt signalling in the carcinogenic process of squamous cell lung carcinoma. PLoS ONE 2013, 8, e57393. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, T.; Malbon, C.C. Structure-function analysis of Frizzleds. Cell. Signal. 2006, 18, 934–941. [Google Scholar] [CrossRef]
- Takada, R.; Hijikata, H.; Kondoh, H.; Takada, S. Analysis of combinatorial effects of Wnts and Frizzleds on beta-catenin/armadillo stabilization and Dishevelled phosphorylation. Genes Cells 2005, 10, 919–928. [Google Scholar] [CrossRef]
- Chen, M.; Zhong, W.; Hu, Y.; Liu, J.; Cai, X. Wnt5a/FZD5/CaMKII signaling pathway mediates the effect of BML-111 on inflammatory reactions in sepsis. Int. J. Clin. Exp. Med. 2015, 8, 17824–17829. [Google Scholar]
- Lu, J.; Zhang, S.; Nakano, H.; Simmons, D.G.; Wang, S.; Kong, S.; Wang, Q.; Shen, L.; Tu, Z.; Wang, W.; et al. A positive feedback loop involving Gcm1 and Fzd5 directs chorionic branching morphogenesis in the placenta. PLoS Biol. 2013, 11, e1001536. [Google Scholar] [CrossRef] [Green Version]
- Chuang, L.S.H.; Ito, Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene 2010, 29, 2605–2615. [Google Scholar] [CrossRef] [Green Version]
- Towle, R.; Truong, D.; Hogg, K.; Robinson, W.P.; Poh, C.F.; Garnis, C. Global analysis of DNA methylation changes during progression of oral cancer. Oral Oncol. 2013, 49, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Ghahhari, N.M.; Babashah, S. Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial–mesenchymal transition in cancer. Eur. J. Cancer 2015, 51, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Semenov, M.V.; Habas, R.; MacDonald, B.T.; He, X. SnapShot: Noncanonical Wnt signaling pathways. Cell 2007, 131, 1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.E.; Beckendorf, S.K. Different Wnt signals act through the Frizzled and RYK receptors during Drosophila salivary gland migration. Development 2007, 134, 2017–2025. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [Green Version]
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jen, W.-C.; De Robertis, E.M.; Kintner, C. The protocadherin PAPC establishes segmental boundaries during somitogenesis in Xenopus embryos. Curr. Biol. 2000, 10, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Tepass, U. Cell sorting in animal development: Signalling and adhesive mechanisms in the formation of tissue boundaries. Curr. Opin. Genet. Dev. 2002, 12, 572–582. [Google Scholar] [CrossRef]
- Zhong, Y.; Brieher, W.M.; Gumbiner, B.M. Analysis of C-cadherin regulation during tissue morphogenesis with an activating antibody. J. Cell Biol. 1999, 144, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Matsunaga, M.; Hatta, K.; Nagafuchi, A.; Takeichi, M. Guidance of optic nerve fibres by N-cadherin adhesion molecules. Nature 1988, 334, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Geisbrecht, E.R.; Montell, D.J. Myosin VI is required for E-cadherin-mediated border cell migration. Nat. Cell Biol. 2002, 4, 616–620. [Google Scholar] [CrossRef]
- Nusrat, A.; Turner, J.R.; Madara, J.L., IV. Regulation of tight junctions by extracellular stimuli: Nutrients, cytokines, and immune cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G851–G857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.-C.; Wang, T.-Y.; Cheng, Y.-A.; Jiang, S.S.; Cheng, C.-W.; Lee, A.Y.-L.; Kao, T.-Y. Expression of E-cadherin, Twist, and p53 and their prognostic value in patients with oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of β-Catenin and E-Cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef]
- Shibamoto, S. Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes. J. Cell Biol. 1995, 128, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Yap, A.S.; Niessen, C.M.; Gumbiner, B.M. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120 ctn. J. Cell Biol. 1998, 141, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Rojas, M.R.; Alcayaga, G.R.; Ramírez, A.M.; Saavedra, J.A.; Rojas, C.; Pinto, A.V.O. Increased nuclear β-catenin expression in oral potentially malignant lesions: A marker of epithelial dysplasia. Med. Oral Patol. Oral Cirugia Bucal 2015, e540–e546. [Google Scholar] [CrossRef]
- Jensen, D.H.; Reibel, J.; Mackenzie, I.C.; Dabelsteen, E. Single cell migration in oral squamous cell carcinoma - possible evidence of epithelial-mesenchymal transition in vivo. J. Oral Pathol. Med. 2015, 44, 674–679. [Google Scholar] [CrossRef]
- Ren, D.; Minami, Y.; Nishita, M. Critical role of Wnt5a-Ror2 signaling in motility and invasiveness of carcinoma cells following Snail-mediated epithelial-mesenchymal transition: Wnt5a-Ror2 signaling in EMT. Genes Cells 2011, 16, 304–315. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldie, K.N.; Wedig, T.; Mitra, A.K.; Aebi, U.; Herrmann, H.; Hoenger, A. Dissecting the 3-D structure of vimentin intermediate filaments by cryo-electron tomography. J. Struct. Biol. 2007, 158, 378–385. [Google Scholar] [CrossRef]
- Aziz, A.; Hess, J.F.; Budamagunta, M.S.; FitzGerald, P.G.; Voss, J.C. Head and Rod 1 interactions in vimentin: Identification of contact sites, structure, and changes with phosphorylation using site-directed spin labeling and electron paramagnetic resonance. J. Biol. Chem. 2009, 284, 7330–7338. [Google Scholar] [CrossRef] [Green Version]
- Rittling, S.R.; Baserga, R. Functional analysis and growth factor regulation of the human vimentin promoter. Mol. Cell Biol. 1987, 7, 3908–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilles, C.; Polette, M.; Mestdagt, M.; Nawrocki-Raby, B.; Ruggeri, P.; Birembaut, P.; Foidart, J.-M. Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res. 2003, 63, 2658–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, B.; He, B.-X.; Cai, J.-H.; Tao, Q.; Lam, A.K. MicroRNA-27a-3p modulates the Wnt/β-Catenin signaling pathway to promote epithelial-mesenchymal transition in oral squamous carcinoma stem cells by targeting SFRP1. Sci. Rep. 2017, 7, 44688. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Li, Q.; Lv, Q.; Wei, Q.; Cao, S.; Gu, J. MiR-27a targets sFRP1 in hFOB cells to regulate proliferation, apoptosis and differentiation. PLoS ONE 2014, 9, e91354. [Google Scholar] [CrossRef]
- Elzi, D.J.; Song, M.; Hakala, K.; Weintraub, S.T.; Shiio, Y. Wnt antagonist SFRP1 functions as a secreted mediator of senescence. Mol. Cell Biol. 2012, 32, 4388–4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, X.; Zhao, C.; Dai, X. Aberrant expression of Wnt antagonist SFRP1 in pancreatic cancer. Chin. Med. J. 2008, 121, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.P.; Melikova, M.; Chuman, Y.; Üren, A.; Baljinnyam, B.; Rubin, J.S. Secreted Frizzled-related protein potentiation versus inhibition of Wnt3a/β-catenin signaling. Cell. Signal. 2014, 26, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E. The Wnt5A/Protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef]
- Su, L.K.; Vogelstein, B.; Kinzler, K.W. Association of the APC tumor suppressor protein with catenins. Science 1993, 262, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Munemitsu, S.; Albert, I.; Souza, B.; Rubinfeld, B.; Polakis, P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl. Acad. Sci. USA 1995, 92, 3046–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shosei, K.; Hideki, Y.; Satoshi, I.; Michiko, K.; Ikuo, S.; Shinya, K.; Akira, K. Axin, a negative regulator of the Wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of β-catenin. J. Biol. Chem. 1998, 273, 10823–10826. [Google Scholar]
- Neufeld, K.L.; White, R.L. Nuclear and cytoplasmic localizations of the adenomatous polyposis coli protein. Proc. Natl. Acad. Sci. USA 1997, 94, 3034–3039. [Google Scholar] [CrossRef] [Green Version]
- Nathke, I.S. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J. Cell Biol. 1996, 134, 165–179. [Google Scholar] [CrossRef]
- Henderson, B.R. Nuclear-cytoplasmic shuttling of APC regulates β-catenin subcellular localization and turnover. Nat. Cell Biol. 2000, 2, 653–660. [Google Scholar] [CrossRef]
- Neufeld, K.L.; Nix, D.A.; Bogerd, H.; Kang, Y.; Beckerle, M.C.; Cullen, B.R.; White, R.L. Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc. Natl. Acad. Sci. USA 2000, 97, 12085–12090. [Google Scholar] [CrossRef] [Green Version]
- Rosin-Arbesfeld, R.; Townsley, F.; Bienz, M. The APC tumour suppressor has a nuclear export function. Nature 2000, 406, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Brocardo, M.G.; Bianchini, M.; Radrizzani, M.; Reyes, G.B.; Dugour, A.V.; Taminelli, G.L.; Gonzalez Solveyra, C.; Santa-Coloma, T.A. APC senses cell–cell contacts and moves to the nucleus upon their disruption. Biochem. Biophys. Res. Commun. 2001, 284, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, C.; Hausmann, G.; Basler, K. β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009, 10, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, R.; Yamamoto, G.; Nagoshi, Y.; Aida, T.; Irie, T.; Tachikawa, T. Expression of adenomatous polyposis coli (APC) in tumorigenesis of human oral squamous cell carcinoma. Oral Oncol. 2004, 40, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liu, X.; Xu, Y.; Zhu, S.; Gao, Y. Co-expression of Axin and APC gene fragments inhibits colorectal cancer cell growth via regulation of the Wnt signaling pathway. Mol. Med. Rep. 2017, 16, 3783–3790. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.; Powell, S.M.; Papadopoulos, N.; Smith, K.J.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Molecular determinants of dysplasia in colorectal lesions. Cancer Res. 1994, 54, 5523–5526. [Google Scholar]
- Sakata, K. Alterations of tumor suppressor genes and the H-ras oncogene in oral squamous cell carcinoma. J. Oral Pathol. Med. 1996, 25, 302–307. [Google Scholar] [CrossRef]
- Kok, S.-H.; Lee, J.-J.; Hsu, H.-C.; Chiang, C.-P.; Kuo, Y.-S.; Kuo, M.Y.-P. Mutations of the adenomatous polyposis coli gene in areca quid and tobacco-associated oral squamous cell carcinomas in Taiwan. J. Oral Pathol. Med. 2002, 31, 395–401. [Google Scholar] [CrossRef]
- Fagman, H.; Larsson, F.; Arvidsson, Y.; Meuller, J.; Nordling, M.; Martinsson, T.; Helmbrecht, K.; Brabant, G.; Nilsson, M. Nuclear accumulation of full-length and truncated adenomatous polyposis coli protein in tumor cells depends on proliferation. Oncogene 2003, 22, 6013–6022. [Google Scholar] [CrossRef] [Green Version]
- Heinen, C.D.; Goss, K.H.; Cornelius, J.R.; Babcock, G.F.; Knudsen, E.S.; Kowalik, T.; Groden, J. The APC tumor suppressor controls entry into S-phase through its ability to regulate the cyclin D/RB pathway. Gastroenterology 2002, 123, 751–763. [Google Scholar] [CrossRef]
- Jaiswal, A.S.; Narayan, S. Zinc stabilizes adenomatous polyposis coli (APC) protein levels and induces cell cycle arrest in colon cancer cells. J. Cell Biochem. 2004, 93, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The mouse Snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell Biol. 2001, 21, 8184–8188. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García de Herreros, A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.-C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-P.; Gao, Z.-L.; Zhou, M.-L.; He, M.-Y.; Xu, X.-H.; Tao, D.-T.; Yang, C.-C.; Liu, L.-K. Snail interacts with Id2 in the regulation of TNF-α-induced cancer cell invasion and migration in OSCC. Am. J. Cancer Res. 2015, 5, 1680–1691. [Google Scholar]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor β-1 induces Snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [Green Version]
- Romano, L.A.; Runyan, R.B. Slug is an essential target of TGFβ2 signaling in the developing chicken heart. Dev. Biol. 2000, 223, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Thuault, S.; Tan, E.-J.; Peinado, H.; Cano, A.; Heldin, C.-H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, L.A. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachelder, R.E.; Yoon, S.-O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial–mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhang, N.; Zhu, J.; Hong, X.-T.; Liu, H.; Ou, Y.-R.; Su, F.; Wang, R.; Li, Y.-M.; Wu, Q. Downregulated connexin32 promotes EMT through the Wnt/β-catenin pathway by targeting Snail expression in hepatocellular carcinoma. Int. J. Oncol. 2017, 50, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Hung, M.-C. Wnt, Hedgehog, and Snail: Sister pathways that control by GSK-3beta and beta-Trcp in the regulation of metastasis. Cell Cycle 2005, 4, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Muratani, M.; Tansey, W.P. How the ubiquitin–proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef]
- Doble, B.W. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-catenin is a target for the ubiquitin–proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Bienz, M.; Clevers, H. Armadillo/β-catenin signals in the nucleus – proof beyond a reasonable doubt? Nat. Cell Biol. 2003, 5, 179–182. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-H.; Chang, J.S.; Hsiao, J.-R.; Yen, Y.-C.; Jiang, S.S.; Liu, S.-H.; Chen, Y.-L.; Shen, Y.-Y.; Chang, J.-Y.; Chen, Y.-W. Tumour cell-derived WNT5B modulates in vitro lymphangiogenesis via induction of partial endothelial-mesenchymal transition of lymphatic endothelial cells. Oncogene 2017, 36, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S. N-Cadherin Expression and Epithelial-Mesenchymal Transition in Pancreatic Carcinoma. Clin. Cancer Res. 2004, 10, 4125–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Shan, W.; Phillips, G.R.; Arndt, K.; Bozdagi, O.; Shapiro, L.; Huntley, G.W.; Benson, D.L.; Colman, D.R. Molecular Modification of N-Cadherin in Response to Synaptic Activity. Neuron 2000, 25, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Uchida, N. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J. Cell Biol. 1996, 135, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelock, M.J.; Johnson, K.R. Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 2003, 19, 207–235. [Google Scholar] [CrossRef]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. A switch from E-Cadherin to N-Cadherin expression indicates epithelial to mesenchymal transition and Is of strong and independent importance for the progress of prostate cancer. Clin. Cancer Res. 2007, 13, 7003–7011. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; van Bokhoven, A.; van Leenders, G.J.; Ruijter, E.T.; Jansen, C.F.; Bussemakers, M.J.; Schalken, J.A. Cadherin switching in human prostate cancer progression. Cancer Res. 2000, 60, 3650–3654. [Google Scholar]
- Sun, H.; Liu, M.; Wu, X.; Yang, C.; Zhang, Y.; Xu, Z.; Gao, K.; Wang, F. Overexpression of N-cadherin and β-catenin correlates with poor prognosis in patients with nasopharyngeal carcinoma. Oncol. Lett. 2017, 13, 1725–1730. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous expression of N-Cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000, 148, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Patel, I.S.; Madan, P.; Getsios, S.; Bertrand, M.A.; MacCalman, C.D. Cadherin switching in ovarian cancer progression. Int. J. Cancer 2003, 106, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Peralta Soler, A.; Knudsen, K.A.; Jaurand, M.-C.; Johnson, K.R.; Wheelock, M.J.; Klein-szanto, A.J.P.; Salazar, H. The differential expression of N-cadherin and E-cadherin distinguishes pleural mesotheliomas from lung adenocarcinomas. Hum. Pathol. 1995, 26, 1363–1369. [Google Scholar] [CrossRef]
- Islam, S. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J. Cell Biol. 1996, 135, 1643–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar]
- Matsunaga, E.; Suzuki, K.; Kato, S.; Kurotani, T.; Kobayashi, K.; Okanoya, K. Dynamic expression of cadherins regulates vocal development in a songbird. PLoS ONE 2011, 6, e25272. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Soeno, Y.; Maeda, G.; Taya, Y.; Aoba, T.; Nasu, M.; Kawashiri, S.; Imai, K. Progression of oral squamous cell carcinoma accompanied with reduced E-Cadherin expression but not cadherin switch. PLoS ONE 2012, 7, e47899. [Google Scholar] [CrossRef]
- DI Domenico, M.; Pierantoni, G.M.; Feola, A.; Esposito, F.; Laino, L.; DE Rosa, A.; Rullo, R.; Mazzotta, M.; Martano, M.; Sanguedolce, F.; et al. Prognostic significance of N-Cadherin expression in oral squamous cell carcinoma. Anticancer Res. 2011, 31, 4211–4218. [Google Scholar]
- Sha Li Jing, J.; Zhaoming, L.; Mingzhi, Z. An essential role for N-cadherin and β-catenin for progression in tongue squamous cell carcinoma and their effect on invasion and metastasis of Tca8113 tongue cancer cells. Oncol. Rep. 2009, 21, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Gasparotto, D.; Polesel, J.; Marzotto, A.; Colladel, R.; Piccinin, S.; Modena, P.; Grizzo, A.; Sulfaro, S.; Serraino, D.; Barzan, L.; et al. Overexpression of TWIST2 correlates with poor prognosis in Head and Neck Squamous Cell Carcinomas. Oncotarget 2011, 2, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Kudo, Y.; Yoshida, M.; Kamata, N.; Ogawa, I.; Takata, T. N-cadherin expression is involved in malignant behavior of head and neck cancer in relation to epithelial-mesenchymal transition. Histol. Histopathol. 2010, 147–156. [Google Scholar] [CrossRef]
- Luo, W.-R.; Wu, A.-B.; Fang, W.-Y.; Li, S.-Y.; Yao, K.-T. Nuclear expression of N-cadherin correlates with poor prognosis of nasopharyngeal carcinoma: Nuclear N-cadherin expression in NPC. Histopathology 2012, 61, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Hulin-Curtis, S.; Williams, H.; Wadey, K.S.; Sala-Newby, G.B.; George, S.J. Targeting Wnt/β-Catenin activated cells with dominant-negative N-cadherin to reduce neointima formation. Mol. Ther. Methods Clin. Dev. 2017, 5, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.; Laskar, S.; Pandey, B.N. Autocrine IL-8 and VEGF mediate epithelial–mesenchymal transition and invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer cells. Cell. Signal. 2013, 25, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 Signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Angela Nieto, M.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, Y.M.; Yang, C.H.; Cho, S.K.; Lee, J.W.; Cho, M. Functional regulation of Slug / Snail2 is dependent on GSK-3β-mediated phosphorylation: GSK-3β-mediated phosphorylation of Slug/Snail2. FEBS J. 2012, 279, 2929–2939. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable -catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Du, X. Crosstalk between tumor cells and microenvironment via Wnt pathway in colorectal cancer dissemination. World J. Gastroenterol. 2008, 14, 1823. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial–mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta BBA Rev. Cancer 2009, 1796, 75–90. [Google Scholar] [CrossRef]
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, A.; Schrauder, M.; Oswald, U.; Knoll, C.; Sellberg, P.; Palmqvist, R.; Niedobitek, G.; Brabletz, T.; Kirchner, T. The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear β-Catenin, Cyclin D1, and p16INK4A and is a region of low proliferation. Am. J. Pathol. 2001, 159, 1613–1617. [Google Scholar] [CrossRef]
- Almangush, A.; Heikkinen, I.; Mäkitie, A.A.; Coletta, R.D.; Läärä, E.; Leivo, I.; Salo, T. Prognostic biomarkers for oral tongue squamous cell carcinoma: A systematic review and meta-analysis. Br. J. Cancer 2017, 117, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Sgaramella, N.; Wilms, T.; Boldrup, L.; Loljung, L.; Gu, X.; Coates, P.; Hassellof, P.; Califano, L.; Lo Muzio, L.; Fahraeus, R.; et al. Ethnicity based variation in expression of E-cadherin in patients with squamous cell carcinoma of the oral tongue. Oncol. Lett. 2018, 16, 6603–6607. [Google Scholar] [CrossRef]
- Zheng, M.; Jiang, Y.; Chen, W.; Li, K.; Liu, X.; Gao, S.; Feng, H.; Wang, S.; Jiang, J.; Ma, X.; et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015, 6, 6794–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Zheng, M.; Jiang, J.; Zhu, G.; Yang, J.; Tang, Y. Hypoxia-inducible factor-1 alpha, in association with TWIST2 and SNIP1, is a critical prognostic factor in patients with tongue squamous cell carcinoma. Oral Oncol. 2011, 47, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Imanishi, Y.; Tomita, T.; Shimoda, M.; Kameyama, K.; Shibata, K.; Sakai, N.; Ozawa, H.; Shigetomi, S.; Fujii, R.; et al. Overexpression of SIP1 and downregulation of E-cadherin predict delayed neck metastasis in stage I/II oral tongue squamous cell carcinoma after partial glossectomy. Ann. Surg. Oncol. 2012, 19, 612–619. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Sha, J.; Kanno, T. The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma. Cancers 2020, 12, 555. https://doi.org/10.3390/cancers12030555
Bai Y, Sha J, Kanno T. The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma. Cancers. 2020; 12(3):555. https://doi.org/10.3390/cancers12030555
Chicago/Turabian StyleBai, Yunpeng, Jingjing Sha, and Takahiro Kanno. 2020. "The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma" Cancers 12, no. 3: 555. https://doi.org/10.3390/cancers12030555
APA StyleBai, Y., Sha, J., & Kanno, T. (2020). The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma. Cancers, 12(3), 555. https://doi.org/10.3390/cancers12030555