The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections


Abstract
:1. Introduction
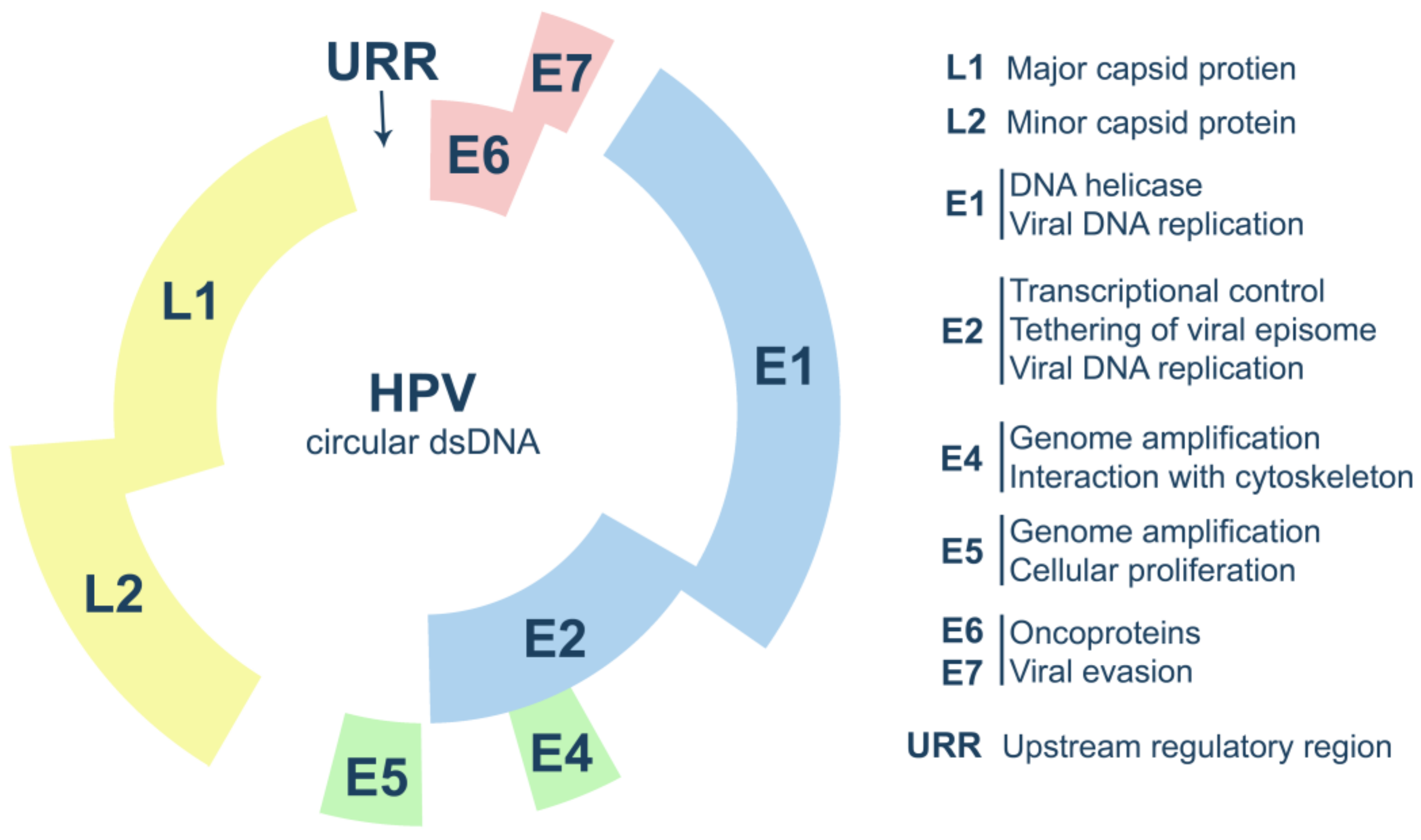
2. Human Papillomavirus Biology
2.1. HPV Life Cycle
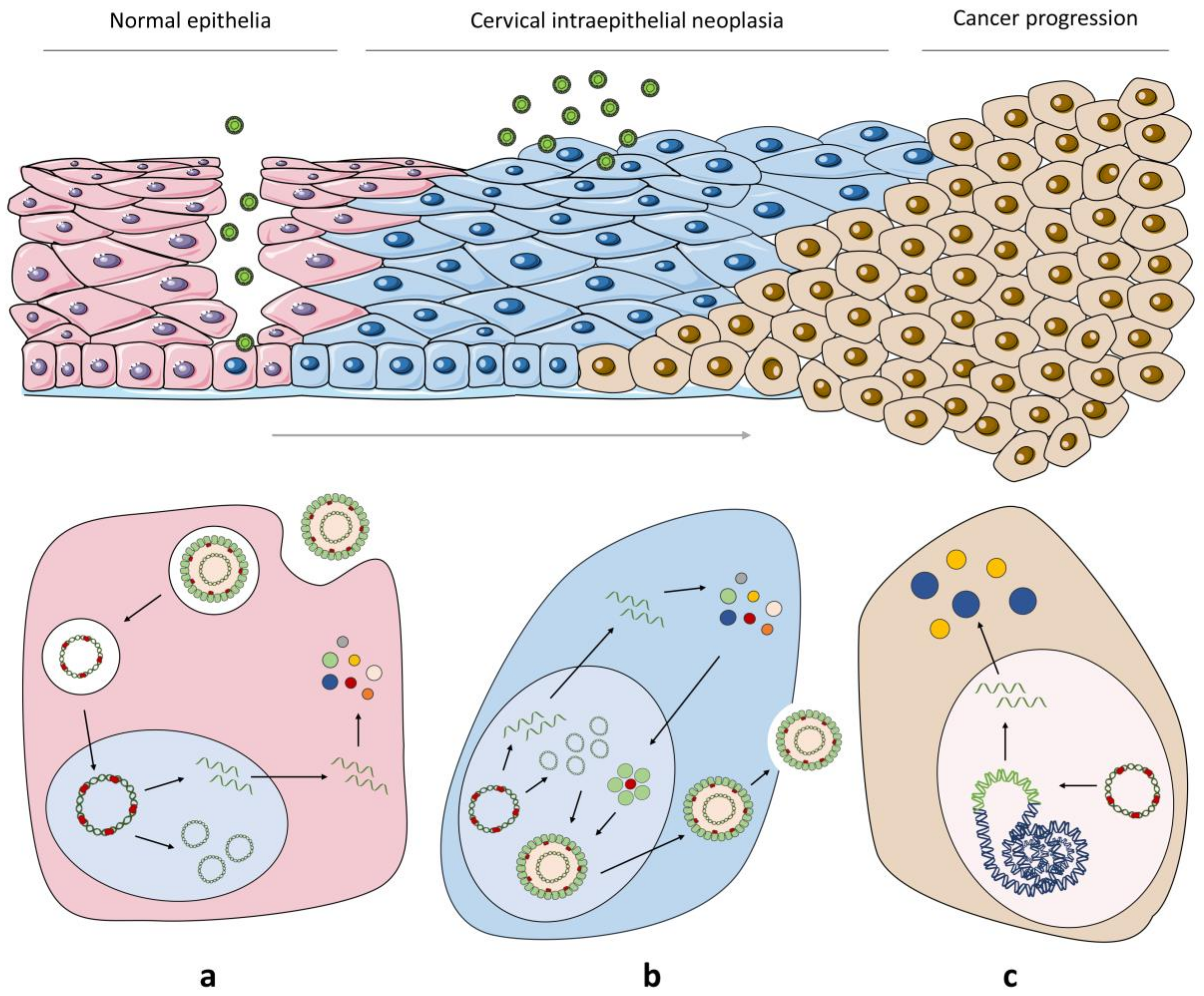
2.2. From HPV Infection to Malignant Transformation
3. Activation of the Antiviral Immune Signalling
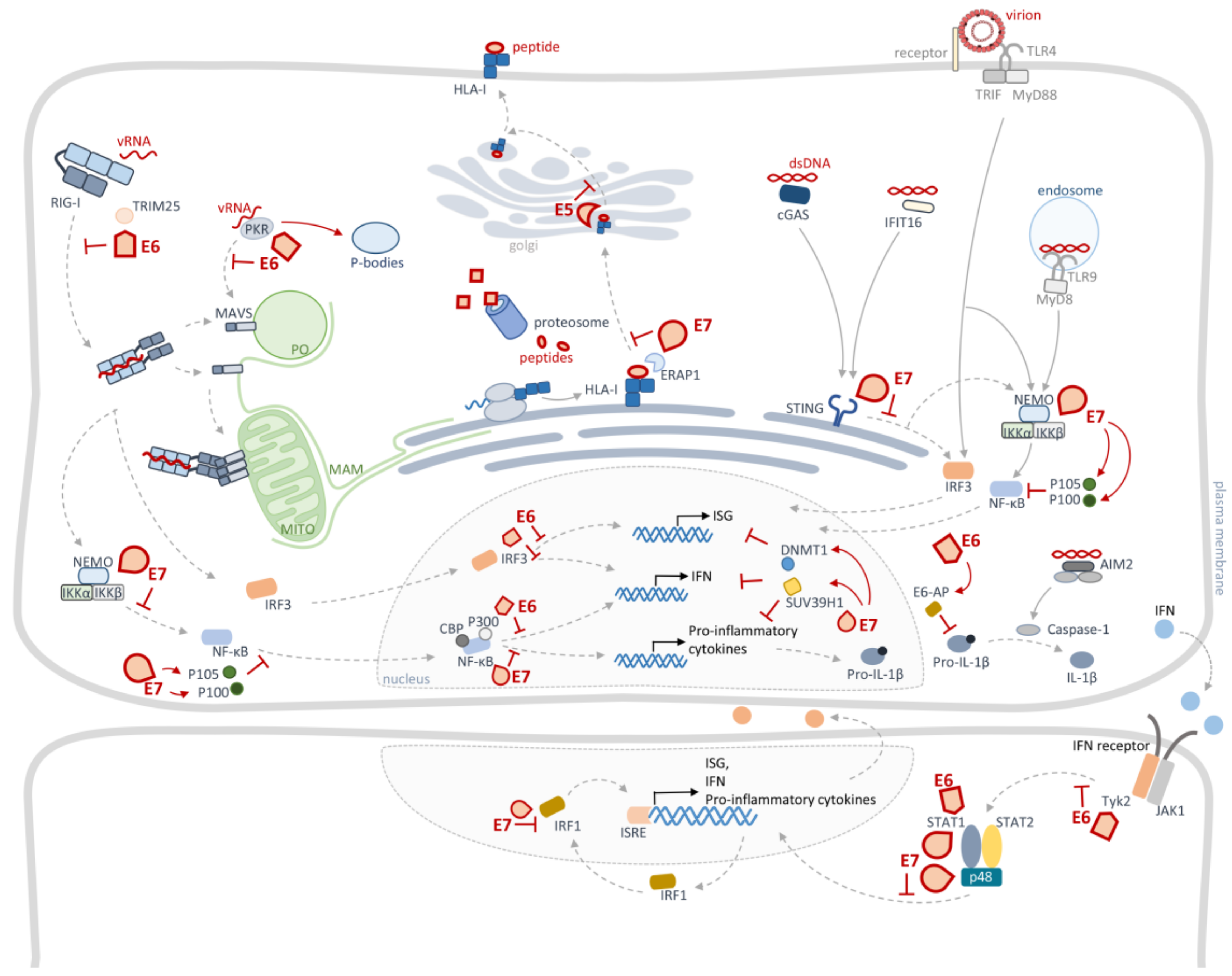
4. Cellular Innate Immunity Evasion by HPV
4.1. HPV Targets Pattern Recognition Receptors
4.2. HPV Targets Interferon Regulatory Factors Signalling
4.3. HPV Targets NF-κBs Signalling
4.4. HPV Targets JAK/STAT Signalling
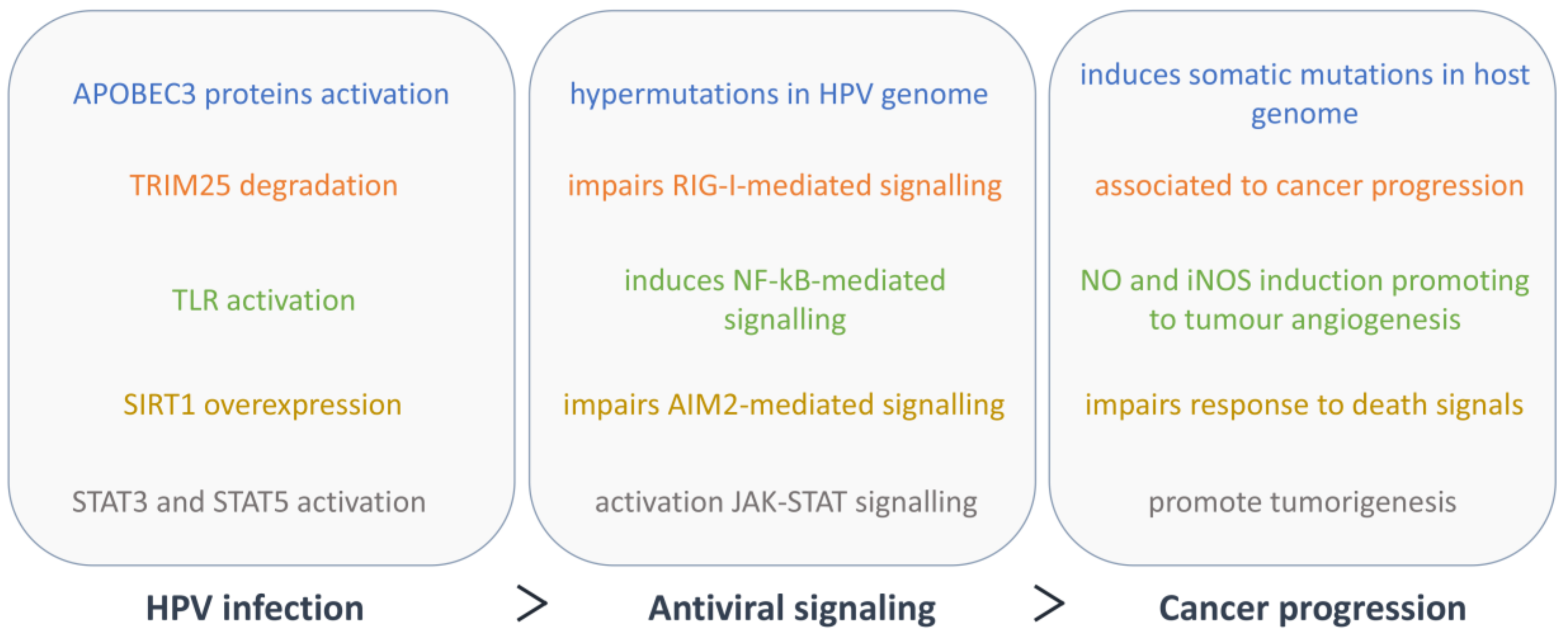
5. Cellular Innate Immunity and Cancer Progression in HPV Infection
6. Conclusions
Funding
Conflicts of Interest
References
- Burd, E.M.; Dean, C.L. Human papillomavirus. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef] [Green Version]
- De Sanjosé, S.; Brotons, M.; Pavón, M.A. The natural history of human papillomavirus infection. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 2–13. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Human Papillomaviruses; International Agency for Research on Cancer: Lyon, France, 2007; Volume 90, ISBN 978-92-832-1290-4. [Google Scholar]
- Medzhitov, R.; Janeway, C.A. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No love lost between viruses and interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, W.M.; Chevillotte, D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [Green Version]
- Woodworth, C.D. HPV innate immunity. Front. Biosci. 2002, 7. [Google Scholar] [CrossRef]
- Harari, A.; Chen, Z.; Burk, R.D. HPV genomics: Past, present and future. Curr. Probl. Dermatol. 2014, 45, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; Mcbride, A.A. The papillomavirus episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, 499–506. [Google Scholar] [CrossRef]
- García-Vallvé, S.; Alonso, A.; Bravo, I.G. Papillomaviruses: Different genes have different histories. Trends Microbiol. 2005, 13, 514–521. [Google Scholar] [CrossRef]
- Egawa, K. Do human papillomaviruses target epidermal stem cells? Dermatology 2003, 207, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousarghin, L.; Touzé, A.; Sizaret, P.-Y.; Coursaget, P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 2003, 77, 3846–3850. [Google Scholar] [CrossRef] [Green Version]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin-and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [Green Version]
- Kämper, N.; Day, P.M.; Nowak, T.; Selinka, H.-C.; Florin, L.; Bolscher, J.; Hilbig, L.; Schiller, J.T.; Sapp, M. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 2006, 80, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Hilbig, L.; Sapp, M. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 2014, 458–459, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kühbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Sapp, M. Human papillomavirus entry: Hiding in a bubble. J. Virol. 2016, 90, 8032–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Teh, B.H.; Tarn, W.Y. A human papillomavirus E2 transcriptional activator: The interactions with cellular splicing factors and potential function in pre-mRNA processing. J. Biol. Chem. 1999, 274, 11832–11841. [Google Scholar] [CrossRef] [Green Version]
- Mole, S.; Milligan, S.G.; Graham, S.V. Human papillomavirus type 16 E2 protein transcriptionally activates the promoter of a key cellular splicing factor, SF2/ASF. J. Virol. 2009, 83, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.V.; Faizo, A.A.A.; Ali Faizo, A.A. Control of human papillomavirus gene expression by alternative splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication—From initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A. Replication and partitioning of papillomavirus genomes. Adv. Virus Res. 2008, 72, 155–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 oncogenes: Key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Ziegert, C.; Wentzensen, N.; Vinokurova, S.; Kisseljov, F.; Einenkel, J.; Hoeckel, M.; Von Knebel Doeberitz, M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003, 22, 3977–3984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-Garau, X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 2006, 25, 5985–5993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammoh, N.; Isaacson, E.; Tomaić, V.; Jackson, D.J.; Doorbar, J.; Banks, L. Inhibition of HPV-16 E7 oncogenic activity by HPV-16 E2. Oncogene 2009, 28, 2299–2304. [Google Scholar] [CrossRef]
- Baldwin, A.; Huh, K.-W.; Munger, K. Human papillomavirus E7 oncoprotein dysregulates steroid receptor coactivator 1 localization and function. J. Virol. 2006, 80, 6669–6677. [Google Scholar] [CrossRef] [Green Version]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk human papillomaviral oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [Green Version]
- Narisawa-Saito, M.; Kiyono, T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of E6 and E7 proteins. Cancer Sci. 2007, 98, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.H.; Yan, L.; Liu, N.; Xu, M.; Cai, H.H. IFI16 promotes cervical cancer progression by upregulating PD-L1 in immunomicroenvironment through STING-TBK1-NF-kB pathway. Biomed. Pharmacother. 2020, 123, 109790. [Google Scholar] [CrossRef] [PubMed]
- Lo Cigno, I.; De Andrea, M.; Borgogna, C.; Albertini, S.; Landini, M.M.; Peretti, A.; Johnson, K.E.; Chandran, B.; Landolfo, S.; Gariglio, M. The nuclear DNA sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J. Virol. 2015, 89, 7506–7520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinholz, M.; Kawakami, Y.; Salzer, S.; Kreuter, A.; Dombrowski, Y.; Koglin, S.; Kresse, S.; Ruzicka, T.; Schauber, J. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch. Dermatol. Res. 2013, 305, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Bahramabadi, R.; Dabiri, S.; Iranpour, M.; Kazemi Arababadi, M. TLR4: An important molecule participating in either anti-human papillomavirus immune responses or development of its related cancers. Viral Immunol. 2019, 32, 417–423. [Google Scholar] [CrossRef]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.G.; Tung, J.-S.; Przysiecki, C.T.; Cook, J.C.; Lehman, E.D.; Sands, J.A.; Jansen, K.U.; Keller, P.M. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 1999, 274, 5810–5822. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Peng, J.; Jabbar, I.A.; Liu, X.; Filgueira, L.; Frazer, I.H.; Thomas, R. Activation of dendritic cells by human papillomavirus-like particles through TLR4 and NF-κB-mediated signalling, moderated by TGF-β. Immunol. Cell Biol. 2005, 83, 83–91. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Daud, I.I.; Scott, M.E.; Ma, Y.; Shiboski, S.; Farhat, S.; Moscicki, A.B. Association between toll-like receptor expression and human papillomavirus type 16 persistence. Int. J. Cancer 2011, 128, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.E.; Ma, Y.; Farhat, S.; Moscicki, A.B. Expression of nucleic acid-sensing toll-like receptors predicts HPV16 clearance associated with an E6-directed cell-mediated response. Int. J. Cancer 2015, 136, 2402–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lembo, D.; Donalisio, M.; De Andrea, M.; Cornaglia, M.; Scutera, S.; Musso, T.; Landolfo, S. A cell-based high-throughput assay for screening inhibitors of human papillomavirus-16 long control region activity. FASEB J. 2006, 20, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; Nakagawa, M.; Moscicki, A.B. Cell-Mediated immune response to human papillomavirus infection. Clin. Diagn. Lab. Immunol. 2001, 8, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowli, S.; Velidandla, R.; Creek, K.E.; Pirisi, L. TGF-β regulation of gene expression at early and late stages of HPV16-mediated transformation of human keratinocytes. Virology 2013, 447, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Lafleur, D.W.; Nardelli, B.; Tsareva, T.; Mather, D.; Feng, P.; Semenuk, M.; Taylor, K.; Buergin, M.; Chinchilla, D.; Roshke, V.; et al. Interferon-κ, a novel type I Interferon expressed in human keratinocytes. J. Biol. Chem. 2001, 276, 39765–39771. [Google Scholar] [CrossRef] [Green Version]
- Woodby, B.L.; Songock, W.K.; Scott, M.L.; Raikhy, G.; Bodily, J.M. Induction of Interferon kappa in human papillomavirus 16 infection by transforming growth factor beta-induced promoter demethylation. J. Virol. 2018, 92, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Habiger, C.; Jäger, G.; Walter, M.; Iftner, T.; Stubenrauch, F. Interferon kappa inhibits human papillomavirus 31 transcription by inducing Sp100 proteins. J. Virol. 2016, 90, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, J.P.; Guétard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.J.; Westrich, J.A.; Van Doorslaer, K.; Pyeon, D. Roles of APOBEC3A and APOBEC3B in human papillomavirus infection and disease progression. Viruses 2017, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Wüstenhagen, E.; Boukhallouk, F.; Negwer, I.; Rajalingam, K.; Stubenrauch, F.; Florin, L. The Myb-related protein MYPOP is a novel intrinsic host restriction factor of oncogenic human papillomaviruses. Oncogene 2018, 37, 6275–6284. [Google Scholar] [CrossRef]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.M.; Miyagi, M.; Wiens, M.E.; Smith, J.G.; Stewart, P. α-Defensin HD5 stabilizes human papillomavirus 16 capsid/core interactions. Pathog. Immun. 2019, 4, 196–234. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.E.; Smith, J.G. Alpha-Defensin HD5 inhibits furin cleavage of human papillomavirus 16 L2 to block infection. J. Virol. 2015, 89, 2866–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, C.B.; Day, P.M.; Thompson, C.D.; Lubkowski, J.; Lu, W.; Lowy, D.R.; Schiller, J.T. Human α-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, P.; Herman, L.; Maillard, C.; Caberg, J.H.; Nikkels, A.; Pierard, G.; Foidart, J.M.; Noel, A.; Boniver, J.; Delvenne, P. Defensins induce the recruitment of dendritic cells in cervical human papillomavirus-associated (pre)neoplastic lesions formed In Vitro and transplanted In Vivo. FASEB J. 2007, 21, 2765–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef]
- Gruener, M.; Bravo, I.G.; Momburg, F.; Alonso, A.; Tomakidi, P. The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol. J. 2007, 4, 116. [Google Scholar] [CrossRef] [Green Version]
- Niebler, M.; Qian, X.; Höfler, D.; Kogosov, V.; Kaewprag, J.; Kaufmann, A.M.; Ly, R.; Böhmer, G.; Zawatzky, R.; Rösl, F.; et al. Post-Translational control of IL-1β via the human papillomavirus type 16 E6 oncoprotein: A novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog. 2013, 9, e1003536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Pauli, E.-K.; Biryukov, J.; Feister, K.F.; Meng, M.; White, E.A.; Münger, K.; Howley, P.M.; Meyers, C.; Gack, M.U. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J. Virol. 2018, 92, e01737-17. [Google Scholar] [CrossRef] [Green Version]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitkovsky, D.; Hehner, S.P.; Hofmann, T.G.; Möller, A.; Schmitz, M.L. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the ikappa B kinase complex. J. Biol. Chem. 2002, 277, 25576–25582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.T.; Pellegrini, S.; Matlashewski, G.J.; Koromilas, A.E. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-α. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef] [Green Version]
- Hebner, C.M.; Wilson, R.; Rader, J.; Bidder, M.; Laimins, L.A. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J. Gen. Virol. 2006, 87, 3183–3193. [Google Scholar] [CrossRef]
- Chang, Y.E.; Laimins, L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-Risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, A.; Winter, J.; Reuschenbach, M.; Blatnik, R.; Klevenz, A.; Bertrand, M.; Hoppe, S.; Von Knebel Doeberitz, M.; Grabowska, A.K.; Riemer, A.B. ERAP1 overexpression in HPV-induced malignancies: A possible novel immune evasion mechanism. Oncoimmunology 2017, 6, e1336594. [Google Scholar] [CrossRef] [Green Version]
- Perea, S.E.; Massimi, P.; Banks, L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int. J. Mol. Med. 2000, 5, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, S.J.; Rhyu, J.W.; Kim, E.J.; Jeon, K.C.; Hwang, E.S.; Park, J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein In Vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Huang, S.-M.; McCance, D.J. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J. Virol. 2002, 76, 8710–8721. [Google Scholar] [CrossRef] [Green Version]
- Barnard, P.; McMillan, N.A.J. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-α. Virology 1999, 259, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; De Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. HPV E7 oncoprotein subverts host innate immunity via SUV39H1-mediated epigenetic silencing of immune sensor genes. J. Virol. 2019, 94, e01812–e018119. [Google Scholar] [CrossRef]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef]
- Pacini, L.; Savini, C.; Ghittoni, R.; Saidj, D.; Lamartine, J.; Hasan, U.A.; Accardi, R.; Tommasino, M. Downregulation of toll-like receptor 9 expression by beta human papillomavirus 38 and implications for cell cycle control. J. Virol. 2015, 89, 11396–11405. [Google Scholar] [CrossRef] [Green Version]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-κB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havard, L.; Rahmouni, S.; Boniver, J.; Delvenne, P. High levels of p105 (NFKB1) and p100 (NFKB2) proteins in HPV16-transformed keratinocytes: Role of E6 and E7 oncoproteins. Virology 2005, 331, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordano, P.; Gillan, V.; Bratlie, S.; Bouvard, V.; Banks, L.; Tommasino, M.; Campo, M.S. The E6E7 oncoproteins of cutaneous human papillomavirus type 38 interfere with the interferon pathway. Virology 2008, 377, 408–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Waterman, P.M.; Jonscher, K.R.; Short, C.M.; Reisdorph, N.A.; Cambier, J.C. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell. Biol. 2008, 28, 5014–5026. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [Green Version]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-Induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Ferreira, A.R.; Ribeiro, D. The interplay between human cytomegalovirus and pathogen recognition receptor signaling. Viruses 2018, 10, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.K.; Wang, Z.; Ban, T.; Yanai, H.; Lu, Y.; Koshiba, R.; Nakaima, Y.; Hangai, S.; Savitsky, D.; Nakasato, M.; et al. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc. Natl. Acad. Sci. USA 2009, 106, 17870–17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchjorsen, J.; Rintahaka, J.; Søby, S.; Horan, K.A.; Poltajainen, A.; Østergaard, L.; Paludan, S.R.; Matikainen, S.; Soby, S.; Horan, K.A.; et al. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.-H.; MacMillian, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.J.; Sparrer, K.M.J.; Van Gent, M.; Lässig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity article. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Magalhães, A.C.; Ferreira, A.R.; Gomes, S.; Vieira, M.; Gouveia, A.; Valença, I.; Islinger, M.; Nascimento, R.; Schrader, M.; Kagan, J.C.; et al. Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep. 2016, 6, 26028. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.R.; Marques, M.; Ribeiro, D. Peroxisomes and innate immunity: Antiviral response and beyond. Int. J. Mol. Sci. 2019, 20, 3795. [Google Scholar] [CrossRef] [Green Version]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic silencing of interferon-κ in human papillomavirus type 16-positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.J.B.; Melief, C.J.M.; et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog. 2013, 9, e1003384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in N-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Richards, K.H.; Wasson, C.W.; Watherston, O.; Doble, R.; Blair, G.E.; Wittmann, M.; Macdonald, A.; Eric Blair, G.; Wittmann, M.; Macdonald, A. The human papillomavirus (HPV) E7 protein antagonises an Imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci. Rep. 2015, 5, 12922. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef]
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 2003, 22, 7871–7881. [Google Scholar] [CrossRef] [Green Version]
- Takami, Y.; Nakagami, H.; Morishita, R.; Katsuya, T.; Cui, T.X.; Ichikawa, T.; Saito, Y.; Hayashi, H.; Kikuchi, Y.; Nishikawa, T.; et al. Ubiquitin carboxyl-terminal hydrolase L1, a novel deubiquitinating enzyme in the vasculature, attenuates NF-κB activation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2184–2190. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Kang, J.W.; Cho, M.C.; Cho, C.W.; Lee, S.J.; Choe, Y.K.; Kim, Y.M.; Choi, I.P.; Park, S.N.; Kim, S.H.; et al. Down modulation of IL-18 expression by human papillomavirus type 16 E6 oncogene via binding to IL-18. FEBS Lett. 2001, 501, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Cho, Y.-S.; Cho, M.-C.; Shim, J.-H.; Lee, K.-A.; Ko, K.-K.; Choe, Y.K.; Park, S.-N.; Hoshino, T.; Kim, S.; et al. Both E6 and E7 oncoproteins of human papillomavirus 16 inhibit IL-18-induced IFN-γ production in human peripheral blood mononuclear and NK cells. J. Immunol. 2001, 167, 497–504. [Google Scholar] [CrossRef] [PubMed]
- So, D.; Shin, H.W.; Kim, J.; Lee, M.; Myeong, J.; Chun, Y.S.; Park, J.W. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene 2018, 37, 5191–5204. [Google Scholar] [CrossRef] [PubMed]
- Senba, M.; Buziba, N.; Mori, N.; Fujita, S.; Morimoto, K.; Wada, A.; Toriyama, K. Human papillomavirus infection induces NF-κB activation in cervical cancer: A comparison with penile cancer. Oncol. Lett. 2011, 2, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmat, N.; Bannazadeh Baghi, H. Association of human papillomavirus infection and inflammation in cervical cancer. Pathog. Dis. 2019, 77, ftz048. [Google Scholar] [CrossRef]
- Barnard, P.; Payne, E.; McMillan, N.A.J. The human papillomavirus E7 protein is able to inhibit the antiviral and anti-growth functions of interferon-α. Virology 2000, 277, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Antonsson, A.; Payne, E.; Hengst, K.; McMillan, N.A.J. The human papillomavirus type 16 E7 protein binds human interferon regulatory factor-9 via a novel PEST domain required for transformation. J. Interf. Cytokine Res. 2006, 26, 455–461. [Google Scholar] [CrossRef]
- Kazemi, S.; Papadopoulou, S.; Li, S.; Su, Q.; Wang, S.; Yoshimura, A.; Matlashewski, G.; Dever, T.E.; Koromilas, A.E. Control of subunit of eukaryotic translation initiation factor 2 (eIF2) phosphorylation by the human papillomavirus type 18 E6 oncoprotein: Implications for eIF2-dependent gene expression and cell death. Mol. Cell. Biol. 2004, 24, 3415–3429. [Google Scholar] [CrossRef] [Green Version]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.J.B.; Melief, C.J.M.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef] [Green Version]
- Morale, M.G.; da Silva Abjaude, W.; Silva, A.M.; Villa, L.L.; Boccardo, E. HPV-Transformed cells exhibit altered HMGB1-TLR4/MyD88-SARM1 signaling axis. Sci. Rep. 2018, 8, 3476. [Google Scholar] [CrossRef] [Green Version]
- Arany, I.; Goel, A.; Tyring, S.K. Interferon response depends on viral transcription in human papillomavirus-containing lesions. Anticancer Res. 1995, 15, 2865–2869. [Google Scholar]
- Martín-Vicente, M.; Medrano, L.M.; Resino, S.; García-Sastre, A.; Martínez, I. TRIM25 in the regulation of the antiviral innate immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Rao, H.; Jin, C.; Liu, J. Involvement of the toll-like receptor/nitric oxide signaling pathway in the pathogenesis of cervical cancer caused by high-risk human papillomavirus infection. Biomed. Res. Int. 2017, 2017, 7830262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimian Rizi, B.; Achreja, A.; Nagrath, D. Nitric oxide: The forgotten child of tumor metabolism. Trends Cancer 2017, 3, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, G.; Verma, G.; Srivastava, Y.; Mehrotra, R.; Das, B.C.; Bharti, A.C. Deregulation of microRNAs Let-7a and miR-21 mediate aberrant STAT3 signaling during human papillomavirus-induced cervical carcinogenesis: Role of E6 oncoprotein. BMC Cancer 2014, 14, 996. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.X.; Zhang, L.; Simayi, D.; Zhang, N.; Tao, L.; Yang, L.; Zhao, J.; Chen, Y.Z.; Li, F.; Zhang, W.J. Human papillomavirus infection correlates with inflammatory stat3 signaling activity and IL-17 level in patients with colorectal cancer. PLoS ONE 2015, 10, e0118391. [Google Scholar] [CrossRef]
- De Andrea, M.; Rittà, M.; Landini, M.M.; Borgogna, C.; Mondini, M.; Kern, F.; Ehrenreiter, K.; Baccarini, M.; Marcuzzi, G.P.; Smola, S.; et al. Keratinocyte-specific stat3 heterozygosity impairs development of skin tumors in human papillomavirus 8 transgenic mice. Cancer Res. 2010, 70, 7938–7948. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant expression and constitutive activation of STAT3 in cervical carcinogenesis: Implications in high-risk human papillomavirus infection. Mol. Cancer 2010, 9, 282. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhang, Y.G.; Sun, J. STAT3 activation in infection and infection-associated cancer. Mol. Cell. Endocrinol. 2017, 451, 80–87. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Farmer, E.; Wu, T.C.; Hung, C.F. Perspectives for therapeutic HPV vaccine development. J. Biomed. Sci. 2016, 23, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Type of Lesions | HPV Types |
|---|---|---|
| High-risk | Intraepithelial neoplasia and cervical cancer | 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 68, 73, 82 (26, 53, 66) |
| Low-risk | Intraepithelial neoplasia or genital warts | 6, 11, 40, 12, 43, 44, 53, 54, 61, 72, 73, 81 |
| Viral Protein | HPV Type | Effects on Innate Immunity |
|---|---|---|
| E2 | HPV16 HPV18 | impairs transcription of the stimulator of IFN genes (STING) and IFN-κ, and their downstream antiviral genes [68] |
| E5 | HPV16 | inhibits the human leukocyte antigen class I (HLA-I) transport, decreasing its surface expression [69,70,71] |
| E6 | HPV16 | induces degradation of pro-IL-β through the ubiquitin ligase E6-AP [72] |
| HPV16 | induces the tripartite motif-containing protein 25 (TRIM25) degradation suppressing the retinoic acid-inducible gene-I (RIG-I)-mediated expression of IFN-β, chemokines, and ISGs [73] | |
| HPV16 | binds to IRF3, inhibiting its transcription activities [74] | |
| HPV16 | reduces transcription activity of CBP/p300, and consequentially NF-κB promoter activity [75,76] | |
| HPV18 | binds to the tyrosine-protein kinase (TYK2), inhibiting the downstream signalling [77] | |
| HPV16 HPV31 | re-localizes the protein kinase R (PKR) to P-bodies, impeding the downstream signalling [78] | |
| HPV31 HPV16 HPV18 | inhibits STAT1 binding and ISGs transcription [79,80] | |
| E7 | HPV16 | induces the overexpression of endoplasmic reticulum aminopeptidase 1 (ERAP1) decreases epitopes presentation [81] |
| HPV16 HPV11 HPV18 | binds to IRF1, inhibiting its promoter activity through histone deacetylases (HDACs) [82,83,84] | |
| HPV16 HPV18 | binds to the NF-κB kinase (IKK) complex, impairing NF-κB signalling [75] | |
| HPV16 | impairs DNA binding activity of NF-κB, through impairment of p65 subunit functions [82,85] | |
| HPV16 | binds to p48, inhibiting the IFN-stimulated gene factor 3 (ISGF3)-mediated gene expression [86] | |
| HPV16 | binds to the DNA methyltransferase (DNMT1), impairing antiviral gene transcription through epigenetic modification [87] | |
| HPV18 | binds to STING, impairing IFNs and pro-inflammatory cytokines expression [88] | |
| HPV18 | induces of the H3K9 methyltransferase (SUV39H1) transcription, which promotes epigenetic silencing of PRRs [89] | |
| HPV16 HPV38 | inhibits of TLR9 promoter region through the formation of a inhibitory complex and induction of HDACs [90,91] | |
| E6 E7 | HPV16 | competes with the transcription promoters of interleukin-18 (IL-18), impairing its expression [85] |
| E6/E7 | HPV16 | inhibit of STAT1 binding to DNA and antiviral genes transcription [92] |
| E6/E7 | HPV16 | induce the overexpression of p100 and p105 and their re-localization, which inhibit the transcriptional activity of NF-κB [93] |
| E6/E7 | HPV38 | decrease MHC-I (human HLA-I) expression through downregulation of STAT1 expression [94] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, A.R.; Ramalho, A.C.; Marques, M.; Ribeiro, D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers 2020, 12, 646. https://doi.org/10.3390/cancers12030646
Ferreira AR, Ramalho AC, Marques M, Ribeiro D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers. 2020; 12(3):646. https://doi.org/10.3390/cancers12030646
Chicago/Turabian StyleFerreira, Ana Rita, Ana Catarina Ramalho, Mariana Marques, and Daniela Ribeiro. 2020. "The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections" Cancers 12, no. 3: 646. https://doi.org/10.3390/cancers12030646
APA StyleFerreira, A. R., Ramalho, A. C., Marques, M., & Ribeiro, D. (2020). The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers, 12(3), 646. https://doi.org/10.3390/cancers12030646