Global RNA Expression and DNA Methylation Patterns in Primary Anaplastic Thyroid Cancer


Abstract
:1. Introduction
2. Results
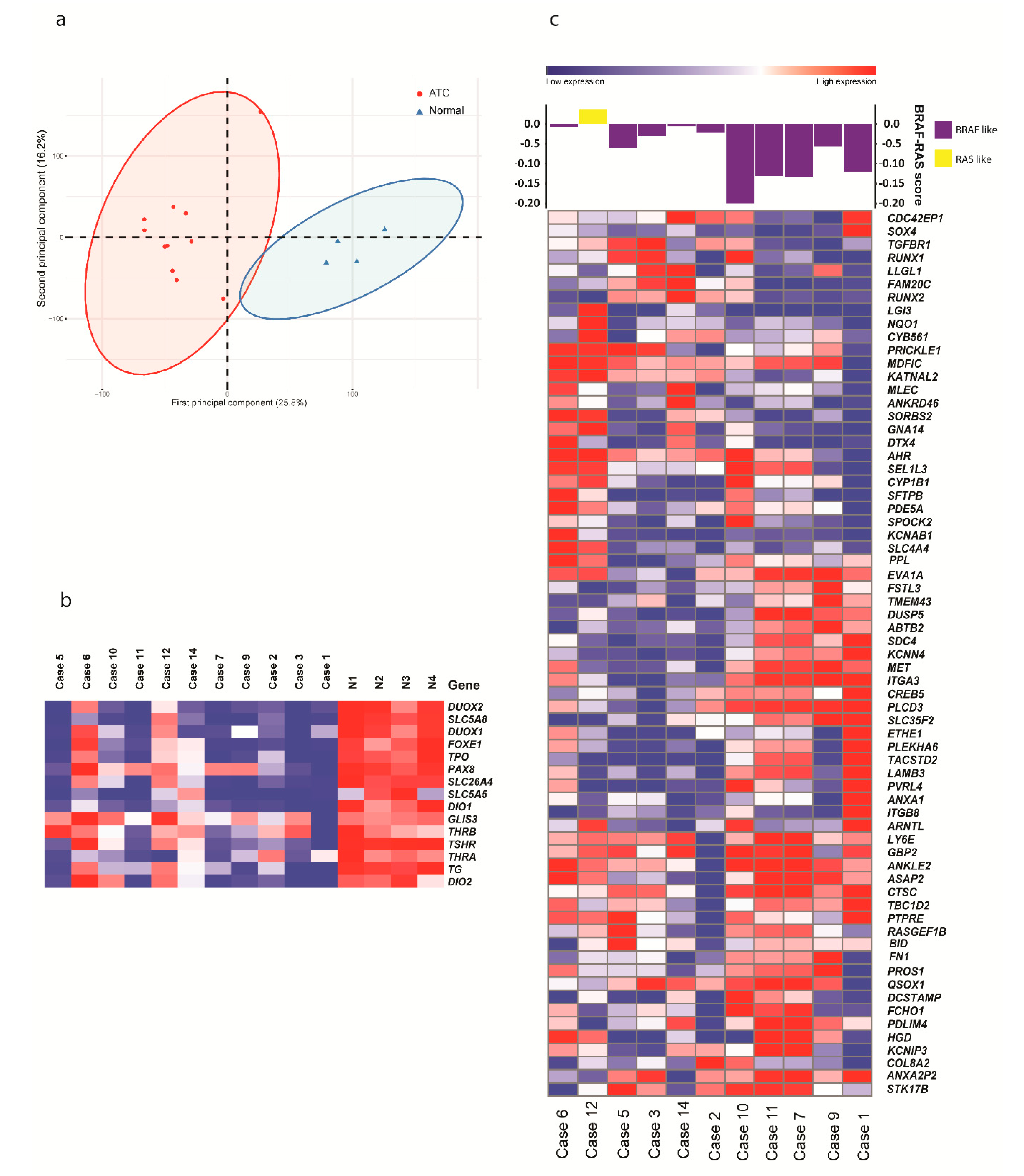
2.1. Global Expression Analysis Shows Upregulation of Cell Cycle Genes and Downregulation of Thyroid-Related Genes
2.2. Methylation Analysis Shows Global Hypomethylation and Hypermethylation of CpG Islands
2.3. Combined Expression and Methylation Analyses Identify Genes Potentially Involved in Tumorigenesis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Expression Analysis
4.3. Methylation Analysis
4.4. Correlation between Methylation and Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roche, A.M.; Fedewa, S.A.; Shi, L.L.; Chen, A.Y. Treatment and survival vary by race/ethnicity in patients with anaplastic thyroid cancer. Cancer 2018, 124, 1780–1790. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Nicolson, N.G.; Choi, J.; Barbieri, A.L.; Kunstman, J.W.; Abou Azar, S.; Knight, J.; Bilguvar, K.; Mane, S.M.; Lifton, R.P.; et al. Clonal evolution analysis of paired anaplastic and well-differentiated thyroid carcinomas reveals shared common ancestor. Genes Chromosomes Cancer 2018, 57, 645–652. [Google Scholar] [CrossRef]
- Capdevila, J.; Mayor, R.; Mancuso, F.M.; Iglesias, C.; Caratu, G.; Matos, I.; Zafon, C.; Hernando, J.; Petit, A.; Nuciforo, P.; et al. Early evolutionary divergence between papillary and anaplastic thyroid cancers. Ann. Oncol. 2018, 29, 1454–1460. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Invest. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, P.; Ponny, S.R.; Xu, H.; Bai, S.; Smallridge, R.; Copland, J.; Sharma, A. Cell cycle M-phase genes are highly upregulated in anaplastic thyroid carcinoma. Thyroid 2017, 27, 236–252. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, G.; Nappi, T.C.; Salerno, P.; Jiang, Y.; Garbi, C.; Ugolini, C.; Miccoli, P.; Basolo, F.; Castellone, M.D.; Cirafici, A.M.; et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007, 67, 10148–10158. [Google Scholar] [CrossRef] [Green Version]
- Hebrant, A.; Dom, G.; Dewaele, M.; Andry, G.; Tresallet, C.; Leteurtre, E.; Dumont, J.E.; Maenhaut, C. mRNA expression in papillary and anaplastic thyroid carcinoma: Molecular anatomy of a killing switch. PLoS ONE 2012, 7, e37807. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Martin-Campos, J.M.; Lerma, E.; Gimenez, G.; Martinez-Guitarte, J.L.; Combalia, N.; Montaner, D.; Matias-Guiu, X.; Dopazo, J.; de Leiva, A.; et al. Molecular profiling related to poor prognosis in thyroid carcinoma. Combining gene expression data and biological information. Oncogene 2008, 27, 1554–1561. [Google Scholar] [CrossRef] [Green Version]
- Pita, J.M.; Figueiredo, I.F.; Moura, M.M.; Leite, V.; Cavaco, B.M. Cell cycle deregulation and TP53 and RAS mutations are major events in poorly differentiated and undifferentiated thyroid carcinomas. J. Clin. Endocrinol. Metab 2014, 99, E497–E507. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Rodero, S.; Fernandez, A.F.; Fernandez-Morera, J.L.; Castro-Santos, P.; Bayon, G.F.; Ferrero, C.; Urdinguio, R.G.; Gonzalez-Marquez, R.; Suarez, C.; Fernandez-Vega, I.; et al. DNA methylation signatures identify biologically distinct thyroid cancer subtypes. J. Clin. Endocrinol. Metab. 2013, 98, 2811–2821. [Google Scholar] [CrossRef]
- Bisarro Dos Reis, M.; Barros-Filho, M.C.; Marchi, F.A.; Beltrami, C.M.; Kuasne, H.; Pinto, C.A.L.; Ambatipudi, S.; Herceg, Z.; Kowalski, L.P.; Rogatto, S.R. Prognostic classifier based on genome-wide DNA methylation profiling in well-differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2017, 102, 4089–4099. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. 2013, 105, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Schagdarsurengin, U.; Gimm, O.; Hoang-Vu, C.; Dralle, H.; Pfeifer, G.P.; Dammann, R. Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res. 2002, 62, 3698–3701. [Google Scholar]
- Schagdarsurengin, U.; Richter, A.M.; Hornung, J.; Lange, C.; Steinmann, K.; Dammann, R.H. Frequent epigenetic inactivation of RASSF2 in thyroid cancer and functional consequences. Mol. Cancer 2010, 9, 264. [Google Scholar] [CrossRef] [Green Version]
- Zuo, H.; Gandhi, M.; Edreira, M.M.; Hochbaum, D.; Nimgaonkar, V.L.; Zhang, P.; Dipaola, J.; Evdokimova, V.; Altschuler, D.L.; Nikiforov, Y.E. Downregulation of Rap1GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. Cancer Res. 2010, 70, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Nakazawa, T.; Ma, D.; Niu, D.; Mochizuki, K.; Kawasaki, T.; Nakamura, N.; Yamane, T.; Kobayashi, M.; Katoh, R. Epigenetic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas. Lab Invest. 2009, 89, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Usadel, H.; Cohen, Y.; Tokumaru, Y.; Guo, Z.; Westra, W.B.; Tong, B.C.; Tallini, G.; Udelsman, R.; Califano, J.A.; et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: A marker of malignancy and a cause of gene silencing. Cancer Res. 2003, 63, 2316–2321. [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Ravi, N.; Yang, M.; Gretarsson, S.; Jansson, C.; Mylona, N.; Sydow, S.R.; Woodward, E.L.; Ekblad, L.; Wennerberg, J.; Paulsson, K. Identification of targetable lesions in anaplastic thyroid cancer by genome profiling. Cancers 2019, 11, 402. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Silva, T.C.; Coetzee, S.G.; Gull, N.; Yao, L.; Hazelett, D.J.; Noushmehr, H.; Lin, D.C.; Berman, B.P. ELMER v.2: An R/Bioconductor package to reconstruct gene regulatory networks from DNA methylation and transcriptome profiles. Bioinformatics 2019, 35, 1974–1977. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Shen, H.; Laird, P.W.; Farnham, P.J.; Berman, B.P. Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol. 2015, 16, 105. [Google Scholar] [CrossRef] [Green Version]
- Klein Hesselink, E.N.; Zafon, C.; Villalmanzo, N.; Iglesias, C.; van Hemel, B.M.; Klein Hesselink, M.S.; Montero-Conde, C.; Buj, R.; Mauricio, D.; Peinado, M.A.; et al. Increased global DNA hypomethylation in distant metastatic and dedifferentiated thyroid cancer. J. Clin. Endocrinol. Metab 2018, 103, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Torano, E.G.; Petrus, S.; Fernandez, A.F.; Fraga, M.F. Global DNA hypomethylation in cancer: Review of validated methods and clinical significance. Clin. Chem. Lab Med. 2012, 50, 1733–1742. [Google Scholar] [CrossRef]
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.J.; Cho, S.W.; Won, J.K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat. Commun. 2019, 10, 2764. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Ljubas, J.; Ovesen, T.; Rusan, M. A Systematic Review of Phase II Targeted Therapy Clinical Trials in Anaplastic Thyroid Cancer. Cancers 2019, 11, 943. [Google Scholar] [CrossRef] [Green Version]
- Bigas, A.; Espinosa, L. The multiple usages of Notch signaling in development, cell differentiation and cancer. Curr. Opin. Cell Biol. 2018, 55, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Burrows, N.; Resch, J.; Cowen, R.L.; von Wasielewski, R.; Hoang-Vu, C.; West, C.M.; Williams, K.J.; Brabant, G. Expression of hypoxia-inducible factor 1 alpha in thyroid carcinomas. Endocr. Relat. Cancer 2010, 17, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, Y.; Chen, X.; Shao, S.; Hu, S.; Zhang, T. MAGI1 mediates tumor metastasis through c-Myb/miR-520h/MAGI1 signaling pathway in renal cell carcinoma. Apoptosis 2019. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Lu, J.; Qu, T.; Feng, Y.; Wang, X.; Liu, C.; Ji, J. MAGI1 inhibits migration and invasion via blocking MAPK/ERK signaling pathway in gastric cancer. Chin. J. Cancer Res. 2017, 29, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaric, J.; Joseph, J.M.; Tercier, S.; Sengstag, T.; Ponsonnet, L.; Delorenzi, M.; Ruegg, C. Identification of MAGI1 as a tumor-suppressor protein induced by cyclooxygenase-2 inhibitors in colorectal cancer cells. Oncogene 2012, 31, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, D.L.; Walker, R.M.; Morris, S.W.; McIntosh, A.M.; Porteous, D.J.; Evans, K.L. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 2016, 9, 22–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Carli, M.M.; Baccarelli, A.A.; Trevisi, L.; Pantic, I.; Brennan, K.J.; Hacker, M.R.; Loudon, H.; Brunst, K.J.; Wright, R.O.; Wright, R.J.; et al. Epigenome-wide cross-tissue predictive modeling and comparison of cord blood and placental methylation in a birth cohort. Epigenomics 2017, 9, 231–240. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Case No. * | Gender | Age | Survival (Months) | Expression Analysis | Methylation Analysis |
|---|---|---|---|---|---|
| 1 | F | 71 | 1 | Yes | Yes |
| 2 | M | 70 | 13 | Yes | Yes |
| 3 | F | 73 | 8 | Yes | Yes |
| 4 | M | 64 | 14 | No | Yes |
| 5 | M | 64 | 4 | Yes | No |
| 6 | F | 72 | 11 | Yes | No |
| 7 | F | 74 | 4 | Yes | Yes |
| 8 | F | 84 | 0 | No | Yes |
| 9 | F | 86 | 1 | Yes | Yes |
| 10 | F | 70 | 18 | Yes | No |
| 11 | M | 84 | 2 | Yes | No |
| 12 | M | 49 | 15 | Yes | Yes |
| 13 | M | 76 | 1 | No | Yes |
| 14 | F | 63 | 7 | Yes | Yes |
| N1 | F | 62 | N/A | Yes | Yes |
| N2 | M | 64 | N/A | Yes | Yes |
| N3 | F | 40 | N/A | Yes | Yes |
| N4 | F | 56 | N/A | Yes | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravi, N.; Yang, M.; Mylona, N.; Wennerberg, J.; Paulsson, K. Global RNA Expression and DNA Methylation Patterns in Primary Anaplastic Thyroid Cancer. Cancers 2020, 12, 680. https://doi.org/10.3390/cancers12030680
Ravi N, Yang M, Mylona N, Wennerberg J, Paulsson K. Global RNA Expression and DNA Methylation Patterns in Primary Anaplastic Thyroid Cancer. Cancers. 2020; 12(3):680. https://doi.org/10.3390/cancers12030680
Chicago/Turabian StyleRavi, Naveen, Minjun Yang, Nektaria Mylona, Johan Wennerberg, and Kajsa Paulsson. 2020. "Global RNA Expression and DNA Methylation Patterns in Primary Anaplastic Thyroid Cancer" Cancers 12, no. 3: 680. https://doi.org/10.3390/cancers12030680
APA StyleRavi, N., Yang, M., Mylona, N., Wennerberg, J., & Paulsson, K. (2020). Global RNA Expression and DNA Methylation Patterns in Primary Anaplastic Thyroid Cancer. Cancers, 12(3), 680. https://doi.org/10.3390/cancers12030680