ATM-Deficient Cancers Provide New Opportunities for Precision Oncology

 ,
,
Abstract
:1. PARP and PARP Inhibitors
2. ATM
3. Targeting ATM-Deficient Cancers
4. ATM Mutation Versus Loss of Function: Identifying Patients Who May Benefit from PARP Inhibitor Treatment
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Ben-Hur, E.; Utsumi, H.; Elkind, M.M. Inhibitors of poly (ADP-ribose) synthesis enhance radiation response by differentially affecting repair of potentially lethal versus sublethal damage. Br. J. Cancer Suppl. 1984, 6, 39–42. [Google Scholar]
- Clark, J.B.; Ferris, G.M.; Pinder, S. Inhibition of nuclear NAD nucleosidase and poly ADP-ribose polymerase activity from rat liver by nicotinamide and 5’-methyl nicotinamide. Biochim. Biophys. Acta 1971, 238, 82–85. [Google Scholar] [CrossRef]
- Tentori, L.; Portarena, I.; Graziani, G. Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharm. Res. 2002, 45, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. Mol. Asp. Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Izhar, L.; Adamson, B.; Ciccia, A.; Lewis, J.; Pontano-Vaites, L.; Leng, Y.; Liang, A.C.; Westbrook, T.F.; Harper, J.W.; Elledge, S.J. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2015, 11, 1486–1500. [Google Scholar] [CrossRef] [Green Version]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef]
- Evers, B.; Drost, R.; Schut, E.; de Bruin, M.; van der Burg, E.; Derksen, P.W.; Holstege, H.; Liu, X.; van Drunen, E.; Beverloo, H.B.; et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin. Cancer Res. 2008, 14, 3916–3925. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.D.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.A.; et al. LBA12_PR—PROfound: Phase III study of olaparib versus enzalutamide or abiraterone for metastatic castration-resistant prostate cancer (mCRPC) with homologous recombination repair (HRR) gene alterations. In Proceedings of the ESMO Congress 2019, Barcelona, Spain, 27 September–1 October 2019. [Google Scholar]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients With Operable Breast Cancer With a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; McDonald, S.; Swift, S.; Turner, N.C.; Ashworth, A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair 2008, 7, 2010–2019. [Google Scholar] [CrossRef]
- Turner, N.C.; Lord, C.J.; Iorns, E.; Brough, R.; Swift, S.; Elliott, R.; Rayter, S.; Tutt, A.N.; Ashworth, A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008, 27, 1368–1377. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteom. MCP 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y. ATM: Expanding roles as a chief guardian of genome stability. Exp. Cell Res. 2014. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. The heterochromatic barrier to DNA double strand break repair: How to get the entry visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Lederman, H.M. Ataxia-telangiectasia (A-T): An emerging dimension of premature ageing. Ageing Res. Rev. 2016. [Google Scholar] [CrossRef]
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem. Sci. 2012, 37, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Tabor, E.; Becker, Y. Abnormal response of ataxia-telangiectasia cells to agents that break the deoxyribose moiety of DNA via a targeted free radical mechanism. Carcinogenesis 1983, 4, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.T.; Bhattarai, T.S.; Schram, A.M.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Chakravarty, D.; Phillips, S.; Kandoth, C.; Penson, A.; et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018, 8, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Gilad, S.; Khosravi, R.; Shkedy, D.; Uziel, T.; Ziv, Y.; Savitsky, K.; Rotman, G.; Smith, S.; Chessa, L.; Jorgensen, T.J.; et al. Predominance of null mutations in ataxia-telangiectasia. Hum. Mol. Genet. 1996, 5, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Greiner, T.C.; Dasgupta, C.; Ho, V.V.; Weisenburger, D.D.; Smith, L.M.; Lynch, J.C.; Vose, J.M.; Fu, K.; Armitage, J.O.; Braziel, R.M.; et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Pro. Natl. Acad. Sci. USA 2006, 103, 2352–2357. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Muzik, H.; Turhan, A.G.; Zamo, A.; O’Connor, M.J.; Bebb, D.G.; Lees-Miller, S.P. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 2010, 9, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, E.; Williamson, C.T.; Ye, R.; Elegbede, A.; Peterson, L.; Lees-Miller, S.P.; Bebb, D.G. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 2014, 13, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl Oncol 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.P.; George, J.; Nowak, M.; Leeser, U.; Buttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, G.; Li, J.; Huang, Y.Y.; Li, Y.; Lin, J.; Chen, L.Z.; Lu, J.P.; Wang, Y.Q.; Wang, C.X.; et al. Association of Tumor Protein p53 and Ataxia-Telangiectasia Mutated Comutation With Response to Immune Checkpoint Inhibitors and Mortality in Patients With Non-Small Cell Lung Cancer. JAMA Netw. Open 2019, 2, e1911895. [Google Scholar] [CrossRef]
- Jette, N.R.; Radhamani, S.; Arthur, G.; Ye, R.; Goutam, S.; Bolyos, A.; Petersen, L.F.; Bose, P.; Bebb, D.G.; Lees-Miller, S.P. Combined poly-ADP ribose polymerase and ataxia-telangiectasia mutated/Rad3-related inhibition targets ataxia-telangiectasia mutated-deficient lung cancer cells. Br. J. Cancer 2019, 121, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Im, S.A.; Jang, H.; Kim, S.; Lee, M.; Kim, D.K.; Yang, Y.; Kim, H.J.; Lee, K.H.; Kim, J.W.; et al. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol. Cancer Ther. 2017, 16, 566–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Saka, B.; Knight, S.; Borges, M.; Childs, E.; Klein, A.; Wolfgang, C.; Herman, J.; Adsay, V.N.; Hruban, R.H.; et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin. Cancer Res. 2014, 20, 1865–1872. [Google Scholar] [CrossRef] [Green Version]
- Jette, N.; Radhamani, S.; Ye, R.; Yu, Y.; Kumar, M.; Arthur, G.; Goutam, S.; Bizmar, T.A.; Bose, P.; Yip, S.; et al. ATM-deficient lung, prostate and pancreatic cancer cells are acutely sensitive to the combination of olaparib and the ATR inhibitor AZD6738. MS ID#: BIORXIV/2020/991166.
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Yusein-Myashkova, S.; Stoykov, I.; Gospodinov, A.; Ugrinova, I.; Pasheva, E. The repair capacity of lung cancer cell lines A549 and H1299 depends on HMGB1 expression level and the p53 status. J. Biochem. 2016, 160, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Lehmann, B.D.; Terrian, D.M.; Abrams, S.L.; Steelman, L.S.; McCubrey, J.A. p53 expression controls prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor Nutlin-3. Cell Cycle 2012, 11, 4579–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Jansma, M.; Linke-Winnebeck, C.; Eustermann, S.; Lammens, K.; Kostrewa, D.; Stakyte, K.; Litz, C.; Kessler, B.; Hopfner, K.P. Near-Complete Structure and Model of Tel1ATM from Chaetomium thermophilum Reveals a Robust Autoinhibited ATP State. Structure 2020, 28, 83–95. [Google Scholar] [CrossRef]
- Yates, L.A.; Williams, R.M.; Hailemariam, S.; Ayala, R.; Burgers, P.; Zhang, X. Cryo-EM Structure of Nucleotide-Bound Tel1(ATM) Unravels the Molecular Basis of Inhibition and Structural Rationale for Disease-Associated Mutations. Structure 2020, 28, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Xin, J.; Xu, Z.; Wang, X.; Tian, Y.; Zhang, Z.; Cai, G. Structural basis of allosteric regulation of Tel1/ATM kinase. Cell Res. 2019, 29, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; de Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2019. [Google Scholar] [CrossRef]
- Bang, Y.J.; Im, S.A.; Lee, K.W.; Cho, J.Y.; Song, E.K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Villaruz, L.C.; Jones, H.; Dacic, S.; Abberbock, S.; Kurland, B.F.; Stabile, L.P.; Siegfried, J.M.; Conrads, T.P.; Smith, N.R.; O’Connor, M.J.; et al. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
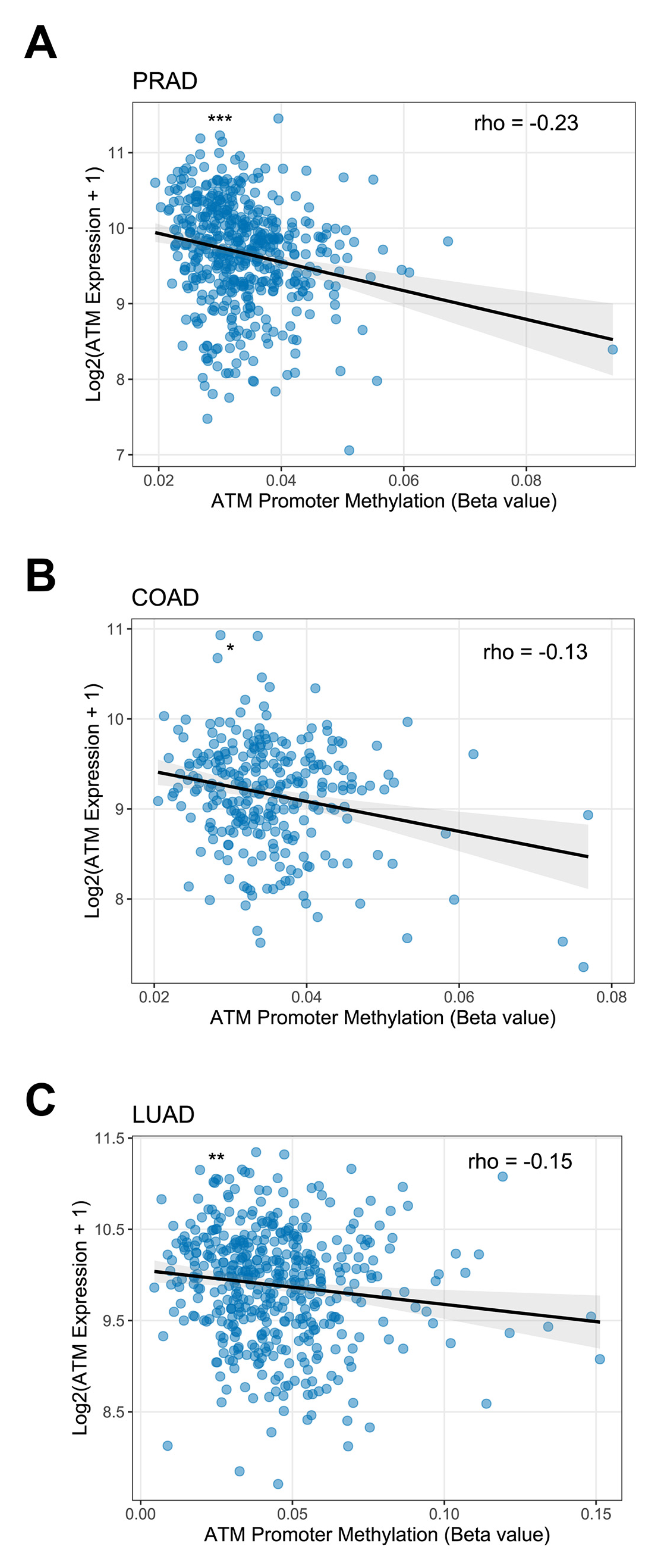
- Kim, W.J.; Vo, Q.N.; Shrivastav, M.; Lataxes, T.A.; Brown, K.D. Aberrant methylation of the ATM promoter correlates with increased radiosensitivity in a human colorectal tumor cell line. Oncogene 2002, 21, 3864–3871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmana, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Zhang, J.; Jing, J.; Dere, R.; Walker, C.L. A new role for ATM in selective autophagy of peroxisomes (pexophagy). Autophagy 2016, 12, 711–712. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoi, N.Y.L.; Choong, C.; Lee, J.; Bellot, G.; Wong, A.L.A.; Goh, B.C.; Pervaiz, S. Targeting Mitochondrial Apoptosis to Overcome Treatment Resistance in Cancer. Cancers 2020, 12, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.L. Modulating Both Tumor Cell Death and Innate Immunity Is Essential for Improving Radiation Therapy Effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, B.; Xie, X.; Xiao, Y.F.; Yang, S.M.; Zhang, J.W. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol. Ther. 2015, 16, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Clinical Trial Number | PARP Inhibitor | ATR Inhibitor | Other Therapy/Status | Cancer Type |
|---|---|---|---|---|
| NCT02723864 | Veliparib/ABT-888 | VX-970 | Cisplatin | Refractory Solid Tumours |
| NCT034R2342 | Olaparib | AZD6738 | Platinum-sensitive or platinum-resistant | Recurrent ovarian cancer (CAPRI trial) |
| NCT03682289 | Olaparib | AZD6738 | None stated | Renal cell carcinoma, urothelial carcinoma, pancreatic cancers and other solid tumours |
| NCT03787680 | Olaparib | AZD6738 | DNA repair proficient/DNA repair deficient | Metastatic Castration-Resistant Prostate Cancer (TRAP trial) |
| NCT04065269 | Olaparib | AZD6738 | ARID1A loss versus no loss | Relapsed gynaecological cancers |
| NCT04267939 | Niraparib | BAY1895344 | Recurrent Advanced Solid Tumours and Ovarian Cancer |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. https://doi.org/10.3390/cancers12030687
Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, Kolinsky M, Williams GJ, Bose P, Lees-Miller SP. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers. 2020; 12(3):687. https://doi.org/10.3390/cancers12030687
Chicago/Turabian StyleJette, Nicholas R., Mehul Kumar, Suraj Radhamani, Greydon Arthur, Siddhartha Goutam, Steven Yip, Michael Kolinsky, Gareth J. Williams, Pinaki Bose, and Susan P. Lees-Miller. 2020. "ATM-Deficient Cancers Provide New Opportunities for Precision Oncology" Cancers 12, no. 3: 687. https://doi.org/10.3390/cancers12030687
APA StyleJette, N. R., Kumar, M., Radhamani, S., Arthur, G., Goutam, S., Yip, S., Kolinsky, M., Williams, G. J., Bose, P., & Lees-Miller, S. P. (2020). ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers, 12(3), 687. https://doi.org/10.3390/cancers12030687