MicroRNAs in Cancer Treatment-Induced Cardiotoxicity



Abstract
:1. Introduction
2. Anticancer Treatment and Cardiotoxicity
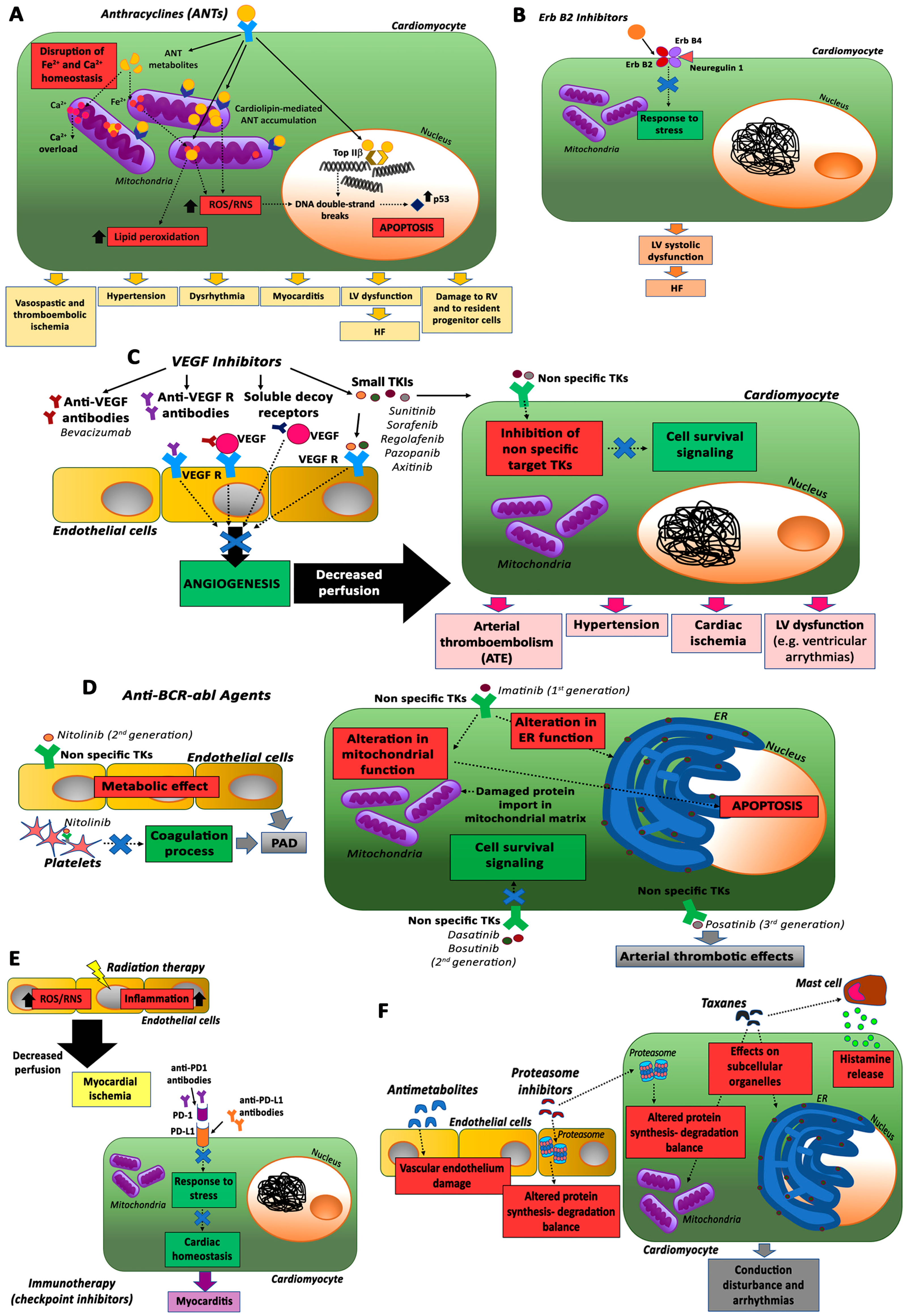
2.1. Anthracyclines (ANTs)
2.2. ErbB2 Inhibitors
2.3. VEGF Inhibitors and Multi-Targeted Kinase Inhibitors
2.4. Anti-BCR-abl Agents
2.5. Immunotherapy and Radiotherapy
2.6. Other Antineoplastic Drugs
3. Monitoring of Cardiotoxicity
4. Role of MicroRNAs in Anti-Cancer Therapy-Induced Cardiotoxicity
4.1. miR-200 Family
4.2. miR-34 Family
4.3. miR-29 Family
4.4. miR-30 Family
4.5. miR-21
4.6. MyomiRs
4.6.1. miR-1
4.6.2. miR-133
4.6.3. miR-208a/b
4.6.4. miR-499
4.7. miR-221/222
4.8. miR-320a
5. Treatment of Cardiotoxicity
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fan, G.-C. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 138–149. [Google Scholar] [PubMed]
- Olson, E.N. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci. Transl. Med. 2014, 6, 239ps3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenhagen, T.; Force, T.; Ewer, M.S.; de Keulenaer, G.W.; Suter, T.M.; Anker, S.D.; Avkiran, M.; de Azambuja, E.; Balligand, J.-L.; Brutsaert, D.L.; et al. Cardiovascular side effects of cancer therapies: A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2011, 13, 1–10. [Google Scholar] [CrossRef]
- Zheng, P.-P.; Li, J.; Kros, J.M. Breakthroughs in modern cancer therapy and elusive cardiotoxicity: Critical research-practice gaps, challenges, and insights. Med. Res. Rev. 2018, 38, 325–376. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Dong, J.; Chen, H. Cardiotoxicity of Anticancer Therapeutics. Front. Cardiovasc. Med. 2018, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Russell, R.R.; Schwartz, R.G.; Panjrath, G.S.; Aronow, W. Cardiac Complications of Cancer Therapy: Pathophysiology, Identification, Prevention, Treatment, and Future Directions. Curr. Cardiol. Rep. 2017, 19, 36. [Google Scholar] [CrossRef]
- Billingham, M.E.; Mason, J.W.; Bristow, M.R.; Daniels, J.R. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer Treat. Rep. 1978, 62, 865–872. [Google Scholar] [PubMed]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments: What the cardiologist needs to know. Nat. Rev. Cardiol. 2010, 7, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Suter, T.M.; Ewer, M.S. Cancer drugs and the heart: Importance and management. Eur. Heart J. 2013, 34, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tocchetti, C.G.; Cadeddu, C.; Di Lisi, D.; Femminò, S.; Madonna, R.; Mele, D.; Monte, I.; Novo, G.; Penna, C.; Pepe, A.; et al. From Molecular Mechanisms to Clinical Management of Antineoplastic Drug-Induced Cardiovascular Toxicity: A Translational Overview. Antioxid. Redox Signal. 2019, 30, 2110–2153. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef]
- Ewer, M.S.; Lippman, S.M. Type II chemotherapy-related cardiac dysfunction: Time to recognize a new entity. J. Clin. Oncol. 2005, 23, 2900–2902. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Y.; Liu, W. Precision cardio-oncology: Understanding the cardiotoxicity of cancer therapy. NPJ Precis. Oncol. 2017, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-M.; Okwuosa, T.M.; Scarabelli, T.; Moudgil, R.; Yeh, E.T.H. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 2. J. Am. Coll. Cardiol. 2017, 70, 2552–2565. [Google Scholar] [CrossRef]
- Smith, L.A.; Cornelius, V.R.; Plummer, C.J.; Levitt, G.; Verrill, M.; Canney, P.; Jones, A. Cardiotoxicity of anthracycline agents for the treatment of cancer: Systematic review and meta-analysis of randomised controlled trials. BMC Cancer 2010, 10, 337. [Google Scholar] [CrossRef] [Green Version]
- Guyot, A.; Ortonne, N.; Valeyrie-Allanore, L.; Bagot, M. Combined Treatment With Rituximab and Anthracycline-Containing Chemotherapy for Primary Cutaneous Large B-Cell Lymphomas, Leg Type, in Elderly Patients. Arch. Dermatol. 2010, 146. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Okuyama, R.; Saida, T.; Uhara, H. Platinum and anthracycline therapy for advanced cutaneous squamous cell carcinoma. Int. J. Clin. Oncol. 2013, 18, 506–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.; Tasaka, H.; Yu, K.P.; Murphy, M.L.; Karnofsky, D.A. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Cancer 1967, 20, 333–353. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadic, M.; Cuspidi, C.; Hering, D.; Venneri, L.; Danylenko, O. The influence of chemotherapy on the right ventricle: Did we forget something? Clin. Cardiol. 2017, 40, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Piegari, E.; De Angelis, A.; Cappetta, D.; Russo, R.; Esposito, G.; Costantino, S.; Graiani, G.; Frati, C.; Prezioso, L.; Berrino, L.; et al. Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res. Cardiol. 2013, 108, 334. [Google Scholar] [CrossRef]
- Piegari, E.; Russo, R.; Cappetta, D.; Esposito, G.; Urbanek, K.; Dell’Aversana, C.; Altucci, L.; Berrino, L.; Rossi, F.; De Angelis, A. MicroRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget 2016, 7, 62312–62326. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Scotti, L.; Franchi, M.; Marchesoni, A.; Corrao, G. Prevalence and incidence of psoriatic arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2018, 48, 28–34. [Google Scholar] [CrossRef]
- Pereira, G.C.; Pereira, S.P.; Tavares, L.C.; Carvalho, F.S.; Magalhães-Novais, S.; Barbosa, I.A.; Santos, M.S.; Bjork, J.; Moreno, A.J.; Wallace, K.B.; et al. Cardiac cytochrome c and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion 2016, 30, 95–104. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresdale, A.R.; Barr, L.H.; Bonow, R.O.; Mathisen, D.J.; Myers, C.E.; Schwartz, D.E.; D’Angelo, T.; Rosenberg, S.A. Prospective randomized study of the role of N-acetyl cysteine in reversing doxorubicin-induced cardiomyopathy. Am. J. Clin. Oncol. 1982, 5, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Hasinoff, B.B.; Patel, D.; Wu, X. The oral iron chelator ICL670A (deferasirox) does not protect myocytes against doxorubicin. Free Radic. Biol. Med. 2003, 35, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.-P.; Azarova, A.M.; Tsai, Y.-C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef] [Green Version]
- Bowles, E.J.A.; Wellman, R.; Feigelson, H.S.; Onitilo, A.A.; Freedman, A.N.; Delate, T.; Allen, L.A.; Nekhlyudov, L.; Goddard, K.A.B.; Davis, R.L.; et al. Risk of heart failure in breast cancer patients after anthracycline and trastuzumab treatment: A retrospective cohort study. J. Natl. Cancer Inst. 2012, 104, 1293–1305. [Google Scholar] [CrossRef]
- Ewer, M.S.; Ewer, S.M. Troponin I provides insight into cardiotoxicity and the anthracycline-trastuzumab interaction. J. Clin. Oncol. 2010, 28, 3901–3904. [Google Scholar] [CrossRef]
- de Korte, M.A.; de Vries, E.G.E.; Lub-de Hooge, M.N.; Jager, P.L.; Gietema, J.A.; van der Graaf, W.T.A.; Sluiter, W.J.; van Veldhuisen, D.J.; Suter, T.M.; Sleijfer, D.T.; et al. 111Indium-trastuzumab visualises myocardial human epidermal growth factor receptor 2 expression shortly after anthracycline treatment but not during heart failure: A clue to uncover the mechanisms of trastuzumab-related cardiotoxicity. Eur. J. Cancer 2007, 43, 2046–2051. [Google Scholar] [CrossRef]
- Gaitanis, G.; Bassukas, I.D. Intralesional bevacizumab as in-add adjuvant to immunocryosurgery for locally advanced basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1117–1121. [Google Scholar] [CrossRef]
- Schutz, F.A.B.; Je, Y.; Azzi, G.R.; Nguyen, P.L.; Choueiri, T.K. Bevacizumab increases the risk of arterial ischemia: A large study in cancer patients with a focus on different subgroup outcomes. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Ranpura, V.; Hapani, S.; Chuang, J.; Wu, S. Risk of cardiac ischemia and arterial thromboembolic events with the angiogenesis inhibitor bevacizumab in cancer patients: A meta-analysis of randomized controlled trials. Acta Oncol. 2010, 49, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Schutz, F.A.B.; Je, Y.; Rosenberg, J.E.; Bellmunt, J. Risk of arterial thromboembolic events with sunitinib and sorafenib: A systematic review and meta-analysis of clinical trials. J. Clin. Oncol. 2010, 28, 2280–2285. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Z.-H.; Qu, X.-J. The adverse effects of sorafenib in patients with advanced cancers. Basic Clin. Pharmacol. Toxicol. 2015, 116, 216–221. [Google Scholar] [CrossRef]
- Chintalgattu, V.; Ai, D.; Langley, R.R.; Zhang, J.; Bankson, J.A.; Shih, T.L.; Reddy, A.K.; Coombes, K.R.; Daher, I.N.; Pati, S.; et al. Cardiomyocyte PDGFR-beta signaling is an essential component of the mouse cardiac response to load-induced stress. J. Clin. Investig. 2010, 120, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moslehi, J.J.; Deininger, M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 4210–4218. [Google Scholar] [CrossRef]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghel, N.; Delgado, D.H.; Lipton, J.H. Cardiovascular toxicities of BCR-ABL tyrosine kinase inhibitors in chronic myeloid leukemia: Preventive strategies and cardiovascular surveillance. Vasc. Health Risk Manag. 2017, 13, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montani, D.; Seferian, A.; Savale, L.; Simonneau, G.; Humbert, M. Drug-induced pulmonary arterial hypertension: A recent outbreak. Eur. Respir. Rev. 2013, 22, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Remsing Rix, L.L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K.L.; Stranzl, T.; Müller, A.; Baumgartner, C.; Valent, P.; Augustin, M.; et al. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 2009, 23, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, T.; Litzow, M.R. Spotlight on ponatinib in the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia: Patient selection and perspectives. Blood Lymphat. Cancer 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, N.E.; Beniata, O.V.; Vitsos, P.; Tsitsilonis, O.; Samara, P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016, 4, 261. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Lahman, M.C.; Chapuis, A.G.; Brownell, I. Immunotherapy for skin cancer. Int. Immunol. 2019, 31, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.R.; Suhag, V.; Semwal, M.; Sharma, N. Radiotherapy: Basic Concepts and Recent Advances. Med. J. Armed Forces India 2010, 66, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Locke, J.; Karimpour, S.; Young, G.; Lockett, M.A.; Perez, C.A. Radiotherapy for epithelial skin cancer. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 748–755. [Google Scholar] [CrossRef]
- Amols, H.I. New technologies in radiation therapy: Ensuring patient safety, radiation safety and regulatory issues in radiation oncology. Health Phys. 2008, 95, 658–665. [Google Scholar] [CrossRef]
- Seibert, J.A. X-ray imaging physics for nuclear medicine technologists. Part 1: Basic principles of X-ray production. J. Nucl. Med. Technol. 2004, 32, 139–147. [Google Scholar] [PubMed]
- Wang, H.; Wei, J.; Zheng, Q.; Meng, L.; Xin, Y.; Yin, X.; Jiang, X. Radiation-induced heart disease: A review of classification, mechanism and prevention. Int. J. Biol. Sci. 2019, 15, 2128–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuomo, J.R.; Sharma, G.K.; Conger, P.D.; Weintraub, N.L. Novel concepts in radiation-induced cardiovascular disease. World J. Cardiol. 2016, 8, 504–519. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.J.M.; Richel, D.J.; van den Brink, R.B.A.; Guchelaar, H.-J. Cardiotoxicity of cytotoxic drugs. Cancer Treat. Rev. 2004, 30, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Eisenhauer, E.A.; Chaudhry, V.; Arbuck, S.G.; Donehower, R.C. Clinical toxicities encountered with paclitaxel (Taxol). Semin. Oncol. 1993, 20, 1–15. [Google Scholar] [PubMed]
- Chong, J.H.; Ghosh, A.K. Coronary Artery Vasospasm Induced by 5-fluorouracil: Proposed Mechanisms, Existing Management Options and Future Directions. Interv. Cardiol. (Lond. Engl.) 2019, 14, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Gavazzoni, M.; Vizzardi, E.; Gorga, E.; Bonadei, I.; Rossi, L.; Belotti, A.; Rossi, G.; Ribolla, R.; Metra, M.; Raddino, R. Mechanism of cardiovascular toxicity by proteasome inhibitors: New paradigm derived from clinical and pre-clinical evidence. Eur. J. Pharmacol. 2018, 828, 80–88. [Google Scholar] [CrossRef]
- Gianni, L.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity in breast cancer patients: Synergism with trastuzumab and taxanes. Cardiovasc. Toxicol. 2007, 7, 67–71. [Google Scholar] [CrossRef]
- Zielinski, C.C. Gemcitabine, anthracycline, and taxane combinations for advanced breast cancer. Oncology (Williston Park) 2003, 17, 36–40. [Google Scholar]
- Spur, E.-M.; Althof, N.; Respondek, D.; Klingel, K.; Heuser, A.; Overkleeft, H.S.; Voigt, A. Inhibition of chymotryptic-like standard proteasome activity exacerbates doxorubicin-induced cytotoxicity in primary cardiomyocytes. Toxicology 2016, 353–354, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Thavendiranathan, P.; Poulin, F.; Lim, K.-D.; Plana, J.C.; Woo, A.; Marwick, T.H. Use of myocardial strain imaging by echocardiography for the early detection of cardiotoxicity in patients during and after cancer chemotherapy: A systematic review. J. Am. Coll. Cardiol. 2014, 63, 2751–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, G.T.; Plana, J.C.; Zhang, N.; Srivastava, D.; Green, D.M.; Ness, K.K.; Daniel Donovan, F.; Metzger, M.L.; Arevalo, A.; Durand, J.-B.; et al. Screening adult survivors of childhood cancer for cardiomyopathy: Comparison of echocardiography and cardiac magnetic resonance imaging. J. Clin. Oncol. 2012, 30, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Miles, D.W. Use of endomyocardial biopsy to assess anthracycline-induced cardiotoxicity. Lancet Oncol. 2005, 6, 67. [Google Scholar] [CrossRef]
- Caspi, O.; Aronson, D. Surviving Cancer without a Broken Heart. Rambam Maimonides Med. J. 2019, 10. [Google Scholar] [CrossRef]
- Frères, P.; Bouznad, N.; Servais, L.; Josse, C.; Wenric, S.; Poncin, A.; Thiry, J.; Moonen, M.; Oury, C.; Lancellotti, P.; et al. Variations of circulating cardiac biomarkers during and after anthracycline-containing chemotherapy in breast cancer patients. BMC Cancer 2018, 18, 102. [Google Scholar] [CrossRef]
- Zhu, J.N.; Fu, Y.H.; Hu, Z.; Li, W.Y.; Tang, C.M.; Fei, H.W.; Yang, H.; Lin, Q.; Gou, D.M.; Wu, S.L.; et al. Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci. Rep. 2017, 7, 11879. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Xia, W.; Hou, M. Macrophage migration inhibitory factor serves a pivotal role in the regulation of radiation-induced cardiac senescencethrough rebalancing the microRNA-34a/sirtuin 1 signaling pathway. Int. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, J.; Zenhausern, F. Emergence of miR-34a in radiation therapy. Crit. Rev. Oncol. Hematol. 2017, 109, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Yee, C.; Lee, K.-M. The effect of radiation on the immune response to cancers. Int. J. Mol. Sci. 2014, 15, 927–943. [Google Scholar] [CrossRef] [Green Version]
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Spallarossa, P.; Altieri, P.; Garibaldi, S.; Ghigliotti, G.; Barisione, C.; Manca, V.; Fabbi, P.; Ballestrero, A.; Brunelli, C.; Barsotti, A. Matrix metalloproteinase-2 and -9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc. Res. 2006, 69, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Eken, S.M.; Christersdottir, T.; Winski, G.; Sangsuwan, T.; Jin, H.; Chernogubova, E.; Pirault, J.; Sun, C.; Simon, N.; Winter, H.; et al. miR-29b Mediates the Chronic Inflammatory Response in Radiotherapy-Induced Vascular Disease. JACC Basic Transl. Sci. 2019, 4, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Salloum, F.N.; Kukreja, R.C. A novel role of microRNA in late preconditioning: Upregulation of endothelial nitric oxide synthase and heat shock protein 70. Circ. Res. 2009, 104, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Cheng, Y.; Yang, J.; Li, J.; Liu, X.; Wang, X.; Wang, D.; Krall, T.J.; Delphin, E.S.; Zhang, C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J. Biol. Chem. 2009, 284, 29514–29525. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Khanna, S.; Hussain, S.-R.A.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 2009, 82, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Viczenczova, C.; Bacova, B.S.; Benova, T.E.; Kura, B.; Yin, C.; Weismann, P.; Kukreja, R.; Slezak, J.; Tribulova, N. Myocardial connexin-43 and PKC signalling are involved in adaptation of the heart to irradiation-induced injury: Implication of miR-1 and miR-21. Gen. Physiol. Biophys. 2016, 35, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Kopcalic, K.; Petrovic, N.; Stanojkovic, T.P.; Stankovic, V.; Bukumiric, Z.; Roganovic, J.; Malisic, E.; Nikitovic, M. Association between miR-21/146a/155 level changes and acute genitourinary radiotoxicity in prostate cancer patients: A pilot study. Pathol. Res. Pract. 2019, 215, 626–631. [Google Scholar] [CrossRef]
- Tang, Y.; Zheng, J.; Sun, Y.; Wu, Z.; Liu, Z.; Huang, G. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int. Heart J. 2009, 50, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.X.; Lin, Q.X.; Fu, Y.H.; Deng, C.Y.; Zhou, Z.L.; Zhu, J.N.; Liu, X.Y.; Zhang, Y.Y.; Li, Y.; Lin, S.G.; et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem. Biophys. Res. Commun. 2009, 381, 597–601. [Google Scholar] [CrossRef]
- Bostjancic, E.; Zidar, N.; Stajner, D.; Glavac, D. MicroRNA miR-1 is up-regulated in remote myocardium in patients with myocardial infarction. Folia Biol. 2010, 56, 27–31. [Google Scholar]
- Tony, H.; Yu, K.; Qiutang, Z. MicroRNA-208a Silencing Attenuates Doxorubicin Induced Myocyte Apoptosis and Cardiac Dysfunction. Oxid. Med. Cell. Longev. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Carvalho, V.; Ferreira, L.R.P.; Bocchi, E.A. Circulating mir-208a fails as a biomarker of doxorubicin-induced cardiotoxicity in breast cancer patients. J. Appl. Toxicol. 2015, 35, 1071–1072. [Google Scholar] [CrossRef]
- Desai, V.G.; C Kwekel, J.; Vijay, V.; Moland, C.L.; Herman, E.H.; Lee, T.; Han, T.; Lewis, S.M.; Davis, K.J.; Muskhelishvili, L.; et al. Early biomarkers of doxorubicin-induced heart injury in a mouse model. Toxicol. Appl. Pharmacol. 2014, 281, 221–229. [Google Scholar] [CrossRef]
- Verjans, R.; Peters, T.; Beaumont, F.J.; van Leeuwen, R.; van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload–Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Zhao, P.; Wang, X.; Wang, J.; Wang, Y.; Song, L.; Zou, Y.; Hui, R. MiR-221 promotes cardiac hypertrophy in vitro through the modulation of p27 expression. J. Cell. Biochem. 2012, 113, 2040–2046. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Watson, C.J.; Gupta, S.K.; O’Connell, E.; Thum, S.; Glezeva, N.; Fendrich, J.; Gallagher, J.; Ledwidge, M.; Grote-Levi, L.; McDonald, K.; et al. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur. J. Heart Fail. 2015, 17, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esplugas, R.; Arenas, M.; Serra, N.; Bellés, M.; Bonet, M.; Gascón, M.; Vallvé, J.-C.; Linares, V. Effect of radiotherapy on the expression of cardiovascular disease-related miRNA-146a, -155, -221 and -222 in blood of women with breast cancer. PLoS ONE 2019, 14, e0217443. [Google Scholar] [CrossRef] [PubMed]
- Plein, A.; Fantin, A.; Ruhrberg, C. Neuropilin regulation of angiogenesis, arteriogenesis, and vascular permeability. Microcirculation 2014, 21, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.M.; Khakoo, A.; Mackey, J.R.; Haykowsky, M.J.; Douglas, P.S.; Jones, L.W. Modulation of anthracycline-induced cardiotoxicity by aerobic exercise in breast cancer: Current evidence and underlying mechanisms. Circulation 2011, 124, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beji, S.; Milano, G.; Scopece, A.; Cicchillitti, L.; Cencioni, C.; Picozza, M.; D’Alessandra, Y.; Pizzolato, S.; Bertolotti, M.; Spaltro, G.; et al. Doxorubicin upregulates CXCR4 via miR-200c/ZEB1-dependent mechanism in human cardiac mesenchymal progenitor cells. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Simůnek, T.; Stérba, M.; Popelová, O.; Adamcová, M.; Hrdina, R.; Gersl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar]
- Damrot, J.; Nübel, T.; Epe, B.; Roos, W.P.; Kaina, B.; Fritz, G. Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. Br. J. Pharmacol. 2006, 149, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative Stress-Induced miR-200c Disrupts the Regulatory Loop among SIRT1, FOXO1, and eNOS. Antioxid. Redox Signal. 2017, 27. [Google Scholar] [CrossRef]
- Potente, M.; Dimmeler, S. Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle 2008, 7, 2117–2122. [Google Scholar] [CrossRef]
- Hu, X.; Liu, H.; Wang, Z.; Hu, Z.; Li, L. miR-200a Attenuated Doxorubicin-Induced Cardiotoxicity through Upregulation of Nrf2 in Mice. Oxid. Med. Cell. Longev. 2019, 2019, 1512326. [Google Scholar] [CrossRef]
- Vacchi-Suzzi, C.; Bauer, Y.; Berridge, B.R.; Bongiovanni, S.; Gerrish, K.; Hamadeh, H.K.; Letzkus, M.; Lyon, J.; Moggs, J.; Paules, R.S.; et al. Perturbation of microRNAs in rat heart during chronic doxorubicin treatment. PLoS ONE 2012, 7, e40395. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-C.; Yang, J.-H.; Liu, G.-H.; Yang, F.; Gong, J.-L.; Jia, M.-G.; Zhang, M.-J.; Zhao, L.-S. miR-34b/c regulates doxorubicin-induced myocardial cell injury through ITCH. Cell Cycle 2019, 18, 3263–3274. [Google Scholar] [CrossRef]
- Cvetković, R.S.; Scott, L.J. Dexrazoxane: A review of its use for cardioprotection during anthracycline chemotherapy. Drugs 2005, 65, 1005–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef]
- Bonfini, L.; Migliaccio, E.; Pelicci, G.; Lanfrancone, L.; Pelicci, P.G. Not all Shc’s roads lead to Ras. Trends. Biochem. Sci. 1996, 21, 257–261. [Google Scholar] [CrossRef]
- Tarrio, M.L.; Grabie, N.; Bu, D.; Sharpe, A.H.; Lichtman, A.H. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J. Immunol. 2012, 188, 4876–4884. [Google Scholar] [CrossRef] [PubMed]
- Baban, B.; Liu, J.Y.; Qin, X.; Weintraub, N.L.; Mozaffari, M.S. Upregulation of Programmed Death-1 and Its Ligand in Cardiac Injury Models: Interaction with GADD153. PLoS ONE 2015, 10, e0124059. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Jing, X.; Yang, J.; Jiang, L.; Chen, J.; Wang, H. MicroRNA-29b Regulates the Mitochondria-Dependent Apoptotic Pathway by Targeting Bax in Doxorubicin Cardiotoxicity. Cell. Physiol. Biochem. 2018, 48, 692–704. [Google Scholar] [CrossRef]
- Leger, K.J.; Leonard, D.; Nielson, D.; de Lemos, J.A.; Mammen, P.P.A.; Winick, N.J. Circulating microRNAs: Potential Markers of Cardiotoxicity in Children and Young Adults Treated With Anthracycline Chemotherapy. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Mehurg, S.M.; Arroyo, J.E.; Stroud, R.E.; DeSantis, S.M.; Spinale, F.G. Relationship between the temporal profile of plasma microRNA and left ventricular remodeling in patients after myocardial infarction. Circ. Cardiovasc. Genet. 2011, 4, 614–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Taylor, N.E.; Lu, L.; Usa, K.; Cowley, A.W.; Ferreri, N.R.; Yeo, N.C.; Liang, M. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension (Dallas Tex. 1979) 2010, 55, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizaki, K.; Ito, R.; Okada, M.; Yoshioka, K.; Uchide, T.; Temma, K.; Mutoh, K.; Uechi, M.; Hara, Y. Enhanced gene expression of myocardial matrix metalloproteinases 2 and 9 after acute treatment with doxorubicin in mice. Pharmacol. Res. 2006, 53, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.-K.T.; Fendler, W.; Chałubińska-Fendler, J.; Acharya, S.S.; O’Leary, C.; Deraska, P.V.; D’Andrea, A.D.; Chowdhury, D.; Kozono, D. Circulating miR-29a and miR-150 correlate with delivered dose during thoracic radiation therapy for non-small cell lung cancer. Radiat. Oncol. 2016, 11, 61. [Google Scholar] [CrossRef]
- Roca-Alonso, L.; Castellano, L.; Mills, A.; Dabrowska, A.F.; Sikkel, M.B.; Pellegrino, L.; Jacob, J.; Frampton, A.E.; Krell, J.; Coombes, R.C.; et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015, 6, e1754. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Chen, J.; Wang, N.; Zhu, G.; Duan, X.; Ling, F. MiRNA-30e mediated cardioprotection of ACE2 in rats with Doxorubicin-induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci. 2017, 169, 69–75. [Google Scholar] [CrossRef]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Zhou, F.; Lu, X.; Zhang, X. Serum miR-30c Level Predicted Cardiotoxicity in Non-small Cell Lung Cancer Patients Treated with Bevacizumab. Cardiovasc. Toxicol. 2018, 18, 284–289. [Google Scholar] [CrossRef]
- Tong, Z.; Jiang, B.; Wu, Y.; Liu, Y.; Li, Y.; Gao, M.; Jiang, Y.; Lv, Q.; Xiao, X. MiR-21 Protected Cardiomyocytes against Doxorubicin-Induced Apoptosis by Targeting BTG2. Int. J. Mol. Sci. 2015, 16, 14511–14525. [Google Scholar] [CrossRef]
- Simone, N.L.; Soule, B.P.; Ly, D.; Saleh, A.D.; Savage, J.E.; Degraff, W.; Cook, J.; Harris, C.C.; Gius, D.; Mitchell, J.B. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS ONE 2009, 4, e6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, J.; Huang, Q.; Wang, L.; Ma, X.; Deng, Q.; Kumar, M.; Zhou, Z.; Li, L.; Zeng, Z.; Young, K.H.; et al. miR-21 depletion in macrophages promotes tumoricidal polarization and enhances PD-1 immunotherapy. Oncogene 2018, 37, 3151–3165. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. MicroRNAs: Redefining Mechanisms in Cardiac Disease. J. Cardiovasc. Pharmacol. 2010, 56, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Cheng, Y.; Tan, N.; Yang, J.; Liu, X.; Cao, X.; He, P.; Dong, X.; Qin, S.; Zhang, C. A translational study of circulating cell-free microRNA-1 in acute myocardial infarction. Clin. Sci. (Lond.) 2010, 119, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Kondo, C.; Morikawa, Y.; Tonomura, Y.; Torii, M.; Yamate, J.; Uehara, T. Plasma miR-208 as a useful biomarker for drug-induced cardiotoxicity in rats. J. Appl. Toxicol. 2015, 35, 173–180. [Google Scholar] [CrossRef]
- Rigaud, V.O.-C.; Ferreira, L.R.P.; Ayub-Ferreira, S.M.; Ávila, M.S.; Brandão, S.M.G.; Cruz, F.D.; Santos, M.H.H.; Cruz, C.B.B.V.; Alves, M.S.L.; Issa, V.S.; et al. Circulating miR-1 as a potential biomarker of doxorubicin-induced cardiotoxicity in breast cancer patients. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kura, B.; Yin, C.; Frimmel, K.; Krizak, J.; Okruhlicova, L.; Kukreja, R.C.; Slezak, J. Changes of microRNA-1, -15b and -21 levels in irradiated rat hearts after treatment with potentially radioprotective drugs. Physiol. Res. 2016, 65 (Suppl. 1), S129–S137. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Shan, H.; Pan, Z.; Li, X.; Li, B.; Xu, C.; Zhang, B.; Zhang, F.; Dong, D.; et al. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: A new mechanism for ischaemic cardioprotection. Cardiovasc. Res. 2009, 84, 434–441. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J. Appl. Physiol. 2007, 102, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Lu, Y.; Pan, Z.; Chu, W.; Luo, X.; Lin, H.; Xiao, J.; Shan, H.; Wang, Z.; Yang, B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007, 120, 3045–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Takahashi, R.; Hiura, Y.; Hirokawa, G.; Fukushima, Y.; Iwai, N. Plasma miR-208 as a Biomarker of Myocardial Injury. Clin. Chem. 2009, 55, 1944–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Q.; Xu, T.; Ding, W.; Zhang, X.; Ji, X.; Yu, T.; Yu, W.; Lin, Z.; Wang, J. miR-499-5p Attenuates Mitochondrial Fission and Cell Apoptosis via p21 in Doxorubicin Cardiotoxicity. Front. Genet. 2018, 9, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. miR221/222 in cancer: Their role in tumor progression and response to therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-specific effects of miR-221/222 in vessels: Molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol. 2012, 52, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Wang, J.; Wang, C.; Wang, X.; Dong, W.; Qiu, W.; Wang, Y.; Zhao, X.; Zou, Y.; Song, L.; et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015, 22, 986–999. [Google Scholar] [CrossRef]
- Lieb, V.; Weigelt, K.; Scheinost, L.; Fischer, K.; Greither, T.; Marcou, M.; Theil, G.; Klocker, H.; Holzhausen, H.-J.; Lai, X.; et al. Serum levels of miR-320 family members are associated with clinical parameters and diagnosis in prostate cancer patients. Oncotarget 2018, 9, 10402–10416. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Bei, Y.; Zhou, Y.; Xiao, J.; Li, X. Non-coding RNAs in cardiac regeneration. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Chakrabarti, S. miR-320 Regulates Glucose-Induced Gene Expression in Diabetes. ISRN Endocrinol. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Zhao, Y.; Li, H.; Yan, M.; Zhou, L.; Chen, C.; Wang, D.W. miR-320a mediates doxorubicin-induced cardiotoxicity by targeting VEGF signal pathway. Aging (Albany. N.Y.) 2016, 8, 192–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambler, G.R.; Johnston, B.M.; Maxwell, L.; Gavin, J.B.; Gluckman, P.D. Improvement of doxorubicin induced cardiomyopathy in rats treated with insulin-like growth factor I. Cardiovasc. Res. 1993, 27, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, W.; Markovich, R.; Chen, J.W.; Wang, P.H. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ. Res. 1998, 83, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menna, P.; Salvatorelli, E. Primary Prevention Strategies for Anthracycline Cardiotoxicity: A Brief Overview. Chemotherapy 2017, 62, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.I.; Martín-Sabroso, C.; Fernández-Carballido, A.; Torres-Suárez, A.I. Current status of nanomedicine in the chemotherapy of breast cancer. Cancer Chemother. Pharmacol. 2019, 84, 689–706. [Google Scholar] [CrossRef]
- Singh, C.K.; Siddiqui, I.A.; El-Abd, S.; Mukhtar, H.; Ahmad, N. Combination chemoprevention with grape antioxidants. Mol. Nutr. Food Res. 2016, 60, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- Kirkham, A.A.; Davis, M.K. Exercise Prevention of Cardiovascular Disease in Breast Cancer Survivors. J. Oncol. 2015, 2015, 917606. [Google Scholar] [CrossRef] [Green Version]
- Herman, E.H.; Ferrans, V.J.; Myers, C.E.; Van Vleet, J.F. Comparison of the effectiveness of (+/-)-1,2-bis(3,5-dioxopiperazinyl-1-yl)propane (ICRF-187) and N-acetylcysteine in preventing chronic doxorubicin cardiotoxicity in beagles. Cancer Res. 1985, 45, 276–281. [Google Scholar]
- Lenčová-Popelová, O.; Jirkovský, E.; Jansová, H.; Jirkovská-Vávrová, A.; Vostatková-Tichotová, L.; Mazurová, Y.; Adamcová, M.; Chládek, J.; Hroch, M.; Pokorná, Z.; et al. Cardioprotective effects of inorganic nitrate/nitrite in chronic anthracycline cardiotoxicity: Comparison with dexrazoxane. J. Mol. Cell. Cardiol. 2016, 91, 92–103. [Google Scholar] [CrossRef]
- Matsui, H.; Morishima, I.; Numaguchi, Y.; Toki, Y.; Okumura, K.; Hayakawa, T. Protective effects of carvedilol against doxorubicin-induced cardiomyopathy in rats. Life Sci. 1999, 65, 1265–1274. [Google Scholar] [CrossRef]
- de Nigris, F.; Rienzo, M.; Schiano, C.; Fiorito, C.; Casamassimi, A.; Napoli, C. Prominent cardioprotective effects of third generation beta blocker nebivolol against anthracycline-induced cardiotoxicity using the model of isolated perfused rat heart. Eur. J. Cancer 2008, 44, 334–340. [Google Scholar] [CrossRef]
- Eindhoven, J.A.; van den Bosch, A.E.; Jansen, P.R.; Boersma, E.; Roos-Hesselink, J.W. The usefulness of brain natriuretic peptide in complex congenital heart disease: A systematic review. J. Am. Coll. Cardiol. 2012, 60, 2140–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglin, M.; Krischer, J.; Tamura, R.; Fink, A.; Bello-Matricaria, L.; McCaskill-Stevens, W.; Munster, P.N. Randomized Trial of Lisinopril Versus Carvedilol to Prevent Trastuzumab Cardiotoxicity in Patients With Breast Cancer. J. Am. Coll. Cardiol. 2019, 73, 2859–2868. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Zhang, J.; Cheng, L.; Wu, X.; Dong, W.; Yang, X.; Li, T.; Liu, X.; Xu, Y.; Li, X.; et al. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2010, 55, 1907–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, A.; Hayward, C.S.; Keogh, A.M.; Kotlyar, E.; McCrohon, J.A.; England, J.F.; Amor, R.; Liu, X.; Li, X.Y.; Zhou, M.D.; et al. Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur. J. Heart Fail. 2011, 13, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Jay, S.M.; Murthy, A.C.; Hawkins, J.F.; Wortzel, J.R.; Steinhauser, M.L.; Alvarez, L.M.; Gannon, J.; Macrae, C.A.; Griffith, L.G.; Lee, R.T. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation 2013, 128, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milano, G.; Biemmi, V.; Lazzarini, E.; Balbi, C.; Ciullo, A.; Bolis, S.; Ameri, P.; Di Silvestre, D.; Mauri, P.; Barile, L.; et al. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| miRNA | Cancer Treatment | Modulation | Tissue/Cells | Source | Ref. |
|---|---|---|---|---|---|
| miR-200c | DOX | up | hCmPC | human | [76] |
| DOX | up | LV heart | mouse | [76] | |
| miR-200a | DOX | down | rat cardiomyocytes | rat | [77] |
| miR-34a | DOX | up | myocardium, plasma, cardiomyocytes | rat | [78] |
| epirubucin | up | plasma | B-cell lymphoma pts | [78] | |
| IR | up | cardiomyocytes | human | [79] | |
| miR-34 b/c | DOX | up | cardiomyocyte cell line | mouse | [80] |
| miR-29b | DOX | down | myocardium, cardiomyocytes | rat | [81] |
| AC | up | plasma | young cancer pts | [82] | |
| IR | down | arteries | human | [83] | |
| IR | down | arteries | ApoE–/– mice | [83] | |
| miR-29a | RT | down | plasma | NSCLC pts | [84] |
| miR-30 family | DOX | down | cardyomyocytes, heart | rat | [85,86] |
| miR-30c | bevacizumab | up | serum | NSCLC pts | [87] |
| miR-21 | DOX | up | myocardium | mouse | [88] |
| DOX | up | cardiomyocytes | rat | [88] | |
| IR | up | fibroblasts | human | [89] | |
| IR | up | myocardium | rat | [90] | |
| RT | up | PBMCs | Prostate cancer pts | [91] | |
| miR-1 | DOX | up | plasma | rat | [92] |
| DOX | up | plasma | Breast cancer pts | [93] | |
| IR | down | myocardium | rat | [90,94] | |
| miR-133a/b | DOX | up | plasma | rat | [95] |
| miR-208a | DOX | up | myocardium | mice | [96] |
| DOX | up | plasma | rat | [96] | |
| DOX | down | myocardium | rat | [97] | |
| miR-208b | DOX | up | myocardium | rat | [97] |
| DOX | up | myocardium | mouse | [98] | |
| miR-499 | DOX | down | myocardium | mouse | [99] |
| DOX | up | serum | mouse | [99] | |
| miR-221/222 | DOX | up | myocardium | mouse | [98] |
| RT | up | plasma | Breast cancer patients | [100] | |
| miR-320a | DOX | up | endothelial cells | human | [101] |
| DOX | up | cardiomyocytes | rat | [101] | |
| AC | down | blood | AML patients | [101] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrini, L.; Sileno, S.; D’Agostino, M.; Foglio, E.; Florio, M.C.; Guzzanti, V.; Russo, M.A.; Limana, F.; Magenta, A. MicroRNAs in Cancer Treatment-Induced Cardiotoxicity. Cancers 2020, 12, 704. https://doi.org/10.3390/cancers12030704
Pellegrini L, Sileno S, D’Agostino M, Foglio E, Florio MC, Guzzanti V, Russo MA, Limana F, Magenta A. MicroRNAs in Cancer Treatment-Induced Cardiotoxicity. Cancers. 2020; 12(3):704. https://doi.org/10.3390/cancers12030704
Chicago/Turabian StylePellegrini, Laura, Sara Sileno, Marco D’Agostino, Eleonora Foglio, Maria Cristina Florio, Vincenzo Guzzanti, Matteo Antonio Russo, Federica Limana, and Alessandra Magenta. 2020. "MicroRNAs in Cancer Treatment-Induced Cardiotoxicity" Cancers 12, no. 3: 704. https://doi.org/10.3390/cancers12030704
APA StylePellegrini, L., Sileno, S., D’Agostino, M., Foglio, E., Florio, M. C., Guzzanti, V., Russo, M. A., Limana, F., & Magenta, A. (2020). MicroRNAs in Cancer Treatment-Induced Cardiotoxicity. Cancers, 12(3), 704. https://doi.org/10.3390/cancers12030704