Renal Tumors of Childhood—A Histopathologic Pattern-Based Diagnostic Approach

 ,
, 

Abstract
:1. Introduction
2. A Pattern-Based Approach to the Use of Ancillary Techniques in Pediatric Renal Tumors
2.1. Epithelial Pattern
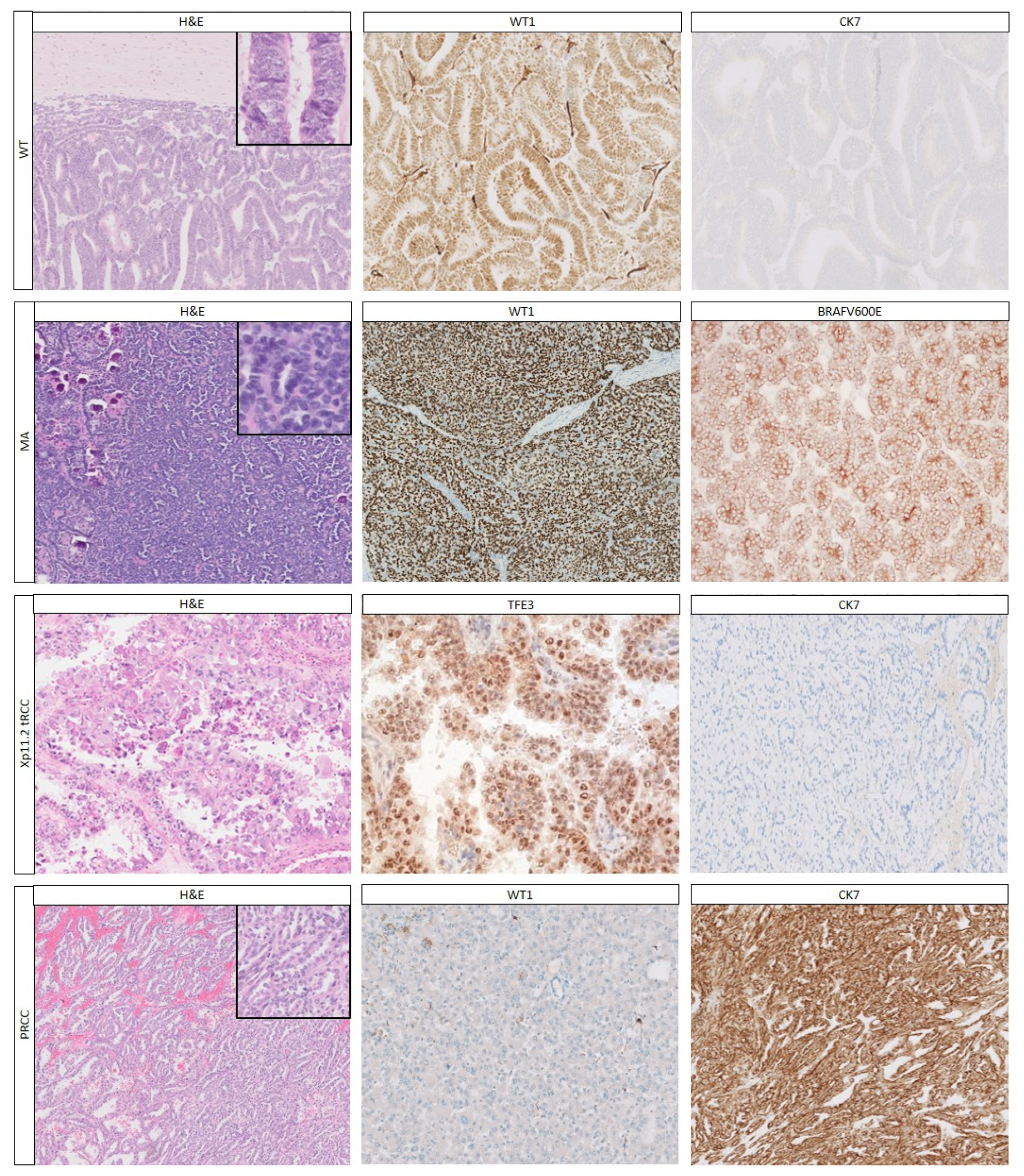
2.1.1. Histology
2.1.2. Immunohistochemistry
2.1.3. Genetics
2.2. Mesenchymal Pattern
2.2.1. Histology
2.2.2. Immunohistochemistry
2.2.3. Genetics
2.3. Undifferentiated Pattern
2.3.1. Histology
2.3.2. Immunohistochemistry
2.3.3. Genetics
2.4. Biphasic Tumors
3. Future Perspectives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pastore, G.; Znaor, A.; Spreafico, F.; Graf, N.; Pritchard-Jones, K.; Steliarova-Foucher, E. Malignant Renal Tumours Incidence and Survival in European Children (1978–1997): Report from the Automated Childhood Cancer Information System Project. Eur. J. Cancer 2006, 42, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel-Eibrink, M.M.; van Tinteren, H.; Rehorst, H.; Coulombe, A.; Patte, C.; de Camargo, B.; de Kraker, J.; Leuschner, I.; Lugtenberg, R.; Pritchard-Jones, K.; et al. Malignant Rhabdoid Tumours of the Kidney (MRTK), Registered on Recent SIOP Protocols from 1993 to 2005: A Report of the SIOP Renal Tumour Study Group. Pediatr. Blood Cancer 2011, 56, 733–737. [Google Scholar] [CrossRef]
- Gooskens, S.L.; Houwing, M.E.; Vujanić, G.M.; Dome, J.S.; Diertens, T.; Coulomb-l’Hermine, A.; Godzinski, J.; Pritchard-Jones, K.; Graf, N.; van den Heuvel-Eibrink, M.M. Congenital Mesoblastic Nephroma 50 Years after Its Recognition: A Narrative Review. Pediatr. Blood Cancer 2017, 64, e26437. [Google Scholar] [CrossRef] [PubMed]
- Wegert, J.; Vokuhl, C.; Ziegler, B.; Ernestus, K.; Leuschner, I.; Furtwangler, R.; Graf, N.; Gessler, M. Tp53 Alterations in Wilms Tumour Represent Progression Events with Strong Intratumour Heterogeneity that Are Closely Linked but Not Limited to Anaplasia. J. Pathol. Clin. Res. 2017, 3, 234–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresswell, G.D.; Apps, J.R.; Chagtai, T.; Mifsud, B.; Bentley, C.C.; Maschietto, M.; Popov, S.D.; Weeks, M.E.; Olsen, Ø.E.; Sebire, N.J.; et al. Intra-Tumor Genetic Heterogeneity in Wilms Tumor: Clonal Evolution and Clinical Implications. EBioMedicine 2016, 9, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.H.; Stiller, C.A.; Walker, L.; Rahman, N. Syndromes and Constitutional Chromosomal Abnormalities Associated with Wilms Tumour. J. Med. Genet. 2006, 43, 705–715. [Google Scholar] [CrossRef]
- Littooij, A.S.; Nikkels, P.G.; Hulsbergen-van de Kaa, C.A.; van de Ven, C.P.; van den Heuvel-Eibrink, M.M.; Olsen, Ø.E. Apparent Diffusion Coefficient as It Relates to Histopathology Findings in Post-Chemotherapy Nephroblastoma: A Feasibility Study. Pediatr. Radiol. 2017, 47, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Van den Heuvel-Eibrink, M.M.; Grundy, P.; Graf, N.; Pritchard-Jones, K.; Bergeron, C.; Patte, C.; van Tinteren, H.; Rey, A.; Langford, C.; Anderson, J.R.; et al. Characteristics and Survival of 750 Children Diagnosed with a Renal Tumor in the First Seven Months of Life: A Collaborative Study by the SIOP/GPOH/SFOP, NWTSG, and UKCCSG Wilms Tumor Study Groups. Pediatr. Blood Cancer 2008, 50, 1130–1134. [Google Scholar] [CrossRef]
- Vujanić, G.M.; Gessler, M.; Ooms, A.H.A.G.; Collini, P.; Coulomb-l’Hermine, A.; D’Hooghe, E.; de Krijger, R.R.; Perotti, D.; Pritchard-Jones, K.; Vokuhl, C.; et al. The UMBRELLA SIOP-RTSG 2016 Wilms Tumour Pathology and Molecular Biology Protocol. Nat. Rev. Urol. 2018, 15, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Jackson, T.J.; Williams, R.D.; Brok, J.; Chowdhury, T.; Ronghe, M.; Powis, M.; Pritchard-Jones, K.; Vujanić, G.M.; Children’s Cancer and Leukaemia Group (CCLG) Renal Tumours Group. The Diagnostic Accuracy and Clinical Utility of Pediatric Renal Tumor Biopsy: Report of the UK Experience in the SIOP UK WT 2001 Trial. Pediatr. Blood Cancer 2019, 66, e27627. [Google Scholar]
- Vujanić, G.M.; Sandstedt, B.; Kelsey, A.; Sebire, N.J. Central Pathology Review in Multicenter Trials and Studies: Lessons from the Nephroblastoma Trials. Cancer 2009, 115, 1977–1983. [Google Scholar] [CrossRef]
- Perlman, E.J. Pediatric Renal Tumors: Practical Updates for the Pathologist. Pediatr. Dev. Pathol. 2005, 8, 320–338. [Google Scholar] [CrossRef]
- Gooskens, S.L.; Furtwangler, R.; Vujanić, G.M.; Dome, J.S.; Graf, N.; van den Heuvel-Eibrink, M.M. Clear Cell Sarcoma of the Kidney: A Review. Eur. J. Cancer 2012, 48, 2219–2226. [Google Scholar] [CrossRef]
- Inamura, K. Translocation Renal Cell Carcinoma: An Update on Clinicopathological and Molecular Features. Cancers 2017, 9, 111. [Google Scholar] [CrossRef]
- Weeks, D.A.; Beckwith, J.B.; Mierau, G.W.; Luckey, D.W. Rhabdoid Tumor of Kidney. A Report of 111 Cases from the National Wilms’ Tumor Study Pathology Center. Am. J. Surg. Pathol. 1989, 13, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Picarsic, J.; Reyes-Mugica, M. Phenotype and Immunophenotype of the Most Common Pediatric Tumors. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Muir, T.E.; Cheville, J.C.; Lager, D.J. Metanephric Adenoma, Nephrogenic Rests, and Wilms’ Tumor: A Histologic and Immunophenotypic Comparison. Am. J. Surg. Pathol. 2001, 25, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Chami, R.; Yin, M.; Marrano, P.; Teerapakpinyo, C.; Shuangshoti, S.; Thorner, P.S. BRAF Mutations in Pediatric Metanephric Tumors. Hum. Pathol. 2015, 46, 1153–1161. [Google Scholar] [CrossRef]
- Charles, A.K.; Mall, S.; Watson, J.; Berry, P.J. Expression of the Wilms’ Tumour Gene WT1 in the Developing Human and in Paediatric Renal Tumours: An Immunohistochemical Study. Mol. Pathol. 1997, 50, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Kinney, S.N.; Eble, J.N.; Hes, O.; Williamson, S.R.; Grignon, D.J.; Wang, M.; Zhang, S.; Baldrige, L.A.; Martignoni, G.; Brunelli, M.; et al. Metanephric Adenoma: The Utility of Immunohistochemical and Cytogenetic Analyses in Differential Diagnosis, including Solid Variant Papillary Renal Cell Carcinoma and Epithelial-Predominant Nephroblastoma. Mod. Pathol. 2015, 28, 1236–1248. [Google Scholar] [CrossRef]
- Sun, Z.; Kan, S.; Zhang, L.; Zhang, Y.; Jing, H.; Huang, G.; Yu, Q.; Wu, J. Immunohistochemical Phenotype and Molecular Pathological Characteristics of Metanephric Adenoma. Int. J. Clin. Exp. Pathol. 2015, 8, 6031–6036. [Google Scholar]
- Truong, L.D.; Shen, S.S. Immunohistochemical Diagnosis of Renal Neoplasms. Arch. Pathol. Lab. Med. 2011, 135, 92–109. [Google Scholar] [PubMed]
- Vasei, M.; Moch, H.; Mousavi, A.; Kajbafzadeh, A.M.; Sauter, G. Immunohistochemical Profiling of Wilms Tumor: A Tissue Microarray Study. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Lal, P.; Hutchinson, B.; Lui, M.Y.; Reuter, V.E.; Ladanyi, M. Aberrant Nuclear Immunoreactivity for TFE3 in Neoplasms with TFE3 Gene Fusions: A Sensitive and Specific Immunohistochemical Assay. Am. J. Surg. Pathol. 2003, 27, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Calio, A.; Eble, J.N.; Hes, O.; Martignoni, G.; Harari, S.E.; Williamson, S.R.; Brunelli, M.; Osunkoya, A.O.; Wang, L.; Comperat, E.; et al. Distinct Clinicopathological Features in Metanephric Adenoma Harboring Braf Mutation. Oncotarget 2017, 8, 54096–54105. [Google Scholar] [CrossRef] [Green Version]
- Udager, A.M.; Pan, J.; Magers, M.J.; Palapattu, G.S.; Morgan, T.M.; Montgomery, J.S.; Weizer, A.Z.; Hafex, K.S.; Miller, D.C.; Wolf, J.S., Jr.; et al. Molecular and Immunohistochemical Characterization Reveals Novel BRAF Mutations in Metanephric Adenoma. Am. J. Surg. Pathol. 2015, 39, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Cajaiba, M.M.; Dyer, L.M.; Geller, J.I.; Jennings, L.J.; George, D.; Kirschmann, D.; Rohan, S.M.; Cost, N.G.; Khanna, G.; Mullen, E.A.; et al. The Classification of Pediatric and Young Adult Renal Cell Carcinomas Registered on the Children’s Oncology Group (COG) Protocol AREN03B2 after Focused Genetic Testing. Cancer 2018, 124, 3381–3389. [Google Scholar] [CrossRef] [Green Version]
- Argani, P.; Hicks, J.; De Marzo, A.M.; Albadine, R.; Illei, P.B.; Ladanyi, M.; Reuter, V.E.; Netto, G.J. Xp11 Translocation Renal Cell Carcinoma (RCC): Extended Immunohistochemical Profile Emphasizing Novel RCC Markers. Am. J. Surg. Pathol. 2010, 34, 1295–1303. [Google Scholar] [CrossRef] [Green Version]
- Martignoni, G.; Gobbo, S.; Camparo, P.; Brunelli, M.; Munari, E.; Segala, D.; Pea, M.; Bonetti, F.; Illei, P.B.; Netto, G.J.; et al. Differential Expression of Cathepsin K in Neoplasms Harboring TFE3 Gene Fusions. Mod. Pathol. 2011, 24, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, J.S.; Malik, F.; Amin, M.B.; Argani, P.; Bahrami, A. MiT Family Translocation Renal Cell Carcinomas: A 15th Anniversary Update. Histol. Histopathol. 2020, 35, 125–136. [Google Scholar]
- Jet Aw, S.; Hong Kuick, C.; Hwee Yong, M.; Wen Quan Lian, D.; Wang, S.; Liang Loh, A.H.; Ling, S.; Lian Peh, G.; Yen Soh, S.; Pheng Loh, A.H.; et al. Novel Karyotypes and Cyclin D1 Immunoreactivity in Clear Cell Sarcoma of the Kidney. Pediatr. Dev. Pathol. 2015, 18, 297–304. [Google Scholar]
- Mirkovic, J.; Calicchio, M.; Fletcher, C.D.; Perez-Atayde, A.R. Diffuse and Strong Cyclin D1 Immunoreactivity in Clear Cell Sarcoma of the Kidney. Histopathology 2015, 67, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Pawel, B.; Szabo, S.; Reyes-Mugica, M.; Timmons, C.; Antonescu, C.R. Diffuse Strong BCOR Immunoreactivity Is a Sensitive and Specific Marker for Clear Cell Sarcoma of the Kidney (CCSK) in Pediatric Renal Neoplasia. Am. J. Surg. Pathol. 2018, 42, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Arva, N.C.; Bonadio, J.; Perlman, E.J.; Cajaiba, M.M. Diagnostic Utility of PAX8, PAX2, and NGFR Immunohistochemical Expression in Pediatric Renal Tumors. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Hill, D.A.; Perlman, E.J. Expression of WT-1, BCL-2, and CD34 by Primary Renal Spindle Cell Tumors in Children. Pediatr. Dev. Pathol. 2004, 7, 577–582. [Google Scholar] [CrossRef]
- Hoot, A.C.; Russo, P.; Judkins, A.R.; Perlman, E.J.; Biegel, J.A. Immunohistochemical Analysis of HSNF5/INI1 Distinguishes Renal and Extra-Renal Malignant Rhabdoid Tumors from Other Pediatric Soft Tissue Tumors. Am. J. Surg. Pathol. 2004, 28, 1485–1491. [Google Scholar] [CrossRef]
- Satoh, F.; Tsutsumi, Y.; Yokoyama, S.; Osamura, R.Y. Comparative Immunohistochemical Analysis of Developing Kidneys, Nephroblastomas and Related Tumors: Considerations on Their Histogenesis. Pathol. Int. 2000, 50, 458–471. [Google Scholar] [CrossRef]
- Argani, P.; Perlman, E.J.; Breslow, N.E.; Browning, N.G.; Green, D.M.; D’Angio, G.J.; Beckwith, J.B. Clear Cell Sarcoma of the Kidney: A Review of 351 Cases from the National Wilms Tumor Study Group Pathology Center. Am. J. Surg. Pathol. 2000, 24, 4–18. [Google Scholar] [CrossRef]
- Wong, M.K.; Ng, C.C.Y.; Kuick, C.H.; Aw, S.J.; Rajasegaran, V.; Lim, J.Q.; Sudhanshi, J.; Loh, E.; Yin, M.; Ma, J.; et al. Clear Cell Sarcomas of Kidney Are Characterized by BCOR Gene Abnormalities Including Exon 15 Internal Tandem Duplications and BCOR-CCNB3 Gene Fusion. Histopathology 2017, 72, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Salvatorelli, L.; Parenti, R.; Leone, G.; Musumeci, G.; Vasquez, E.; Magro, G. Wilms Tumor 1 (WT1) Protein: Diagnostic Utility in Pediatric Tumors. Acta Histochem. 2015, 117, 367–378. [Google Scholar] [CrossRef]
- El Demellawy, D.; Cundiff, C.A.; Nasr, A.; Ozolek, J.A.; Elawabdeh, N.; Caltharp, S.A.; Masoudian, P.; Sullivan, K.J.; de Nanassy, J.; Shehata, B.M. Congenital Mesoblastic Nephroma: A Study of 19 Cases Using Immunohistochemistry and ETRV6-NTRK3 Fusion Gene Rearrangement. Pathology 2016, 48, 47–50. [Google Scholar] [CrossRef]
- Argani, P.; Beckwith, J.B. Metanephric Stromal Tumor: Report of 31 Cases of a Distinctive Pediatric Renal Neoplasm. Am. J. Surg. Pathol. 2000, 24, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.W.; Qu, F.; Hu, S.W.; Zheng, J.Y.; Wang, J.M.; Zhu, X.Y.; Li, J.; Guo, H.Q. Metanephric Adenofibroma in a 10-Year-Old Boy: Report of a Case and Review of the Literature. Int. J. Clin. Exp. Pathol. 2015, 8, 3250–3256. [Google Scholar] [PubMed]
- Arnold, M.A.; Schoenfield, L.; Limketkai, B.N.; Arnold, C.A. Diagnostic Pitfalls of Differentiating Desmoplastic Small Round Cell Tumor (DSRCT) from Wilms Tumor (WT): Overlapping Morphologic and Immunohistochemical Features. Am. J. Surg. Pathol. 2014, 38, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, R.E.; Folpe, A.L.; Lapham, R.L.; Ro, J.Y.; O’Shea, P.A.; Weiss, S.W.; Amin, M.B. Primary Ewing’s Sarcoma/Primitive Neuroectodermal Tumor of the Kidney: A Clinicopathologic and Immunohistochemical Analysis of 11 Cases. Am. J. Surg. Pathol. 2002, 26, 320–327. [Google Scholar] [CrossRef]
- Ramani, P.; Cowell, J.K. The Expression Pattern of Wilms’ Tumour Gene (WT1) Product in Normal Tissues and Paediatric Renal Tumours. J. Pathol. 1996, 179, 162–168. [Google Scholar] [CrossRef]
- Magro, G.; Longo, F.R.; Angelico, G.; Spadola, S.; Amore, F.F.; Salvatorelli, L. Immunohistochemistry as Potential Diagnostic Pitfall in the Most Common Solid Tumors of Children and Adolescents. Acta Histochem. 2015, 117, 397–414. [Google Scholar] [CrossRef]
- Barnoud, R.; Sabourin, J.C.; Pasquier, D.; Ranchere, D.; Bailly, C.; Terrier-Lacombe, M.J.; Pasquier, B. Immunohistochemical Expression of WT1 by Desmoplastic Small Round Cell Tumor: A Comparative Study with Other Small Round Cell Tumors. Am. J. Surg. Pathol. 2000, 24, 830–836. [Google Scholar] [CrossRef]
- Thomas, J.O.; Nijjar, J.; Turley, H.; Micklem, K.; Gatter, K.C. NB84: A New Monoclonal Antibody for the Recognition of Neuroblastoma in Routinely Processed Material. J. Pathol. 1991, 163, 69–75. [Google Scholar] [CrossRef]
- Hung, Y.P.; Lee, J.P.; Bellizzi, A.M.; Hornick, J.L. PHOX2b Reliably Distinguishes Neuroblastoma among Small Round Blue Cell Tumours. Histopathology 2017, 71, 786–794. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Alaggio, R.; D’Agata, V.; Nicoletti, F.; Di Cataldo, A.; Parenti, R. Diagnostic Utility of Cyclin D1 in the Diagnosis of Small Round Blue Cell Tumors in Children and Adolescents. Hum. Pathol. 2017, 60, 58–65. [Google Scholar] [CrossRef]
- Agaimy, A. The Expanding Family of SMARCB1 (INI1)-Deficient Neoplasia: Implications of Phenotypic, Biological, and Molecular Heterogeneity. Adv. Anat. Pathol. 2014, 21, 394–410. [Google Scholar] [CrossRef]
- Hachitanda, Y.; Tsuneyoshi, M.; Enjoji, M. Expression of Pan-Neuroendocrine Proteins in 53 Neuroblastic Tumors. An Immunohistochemical Study with Neuron-Specific Enolase, Chromogranin, and Synaptophysin. Arch. Pathol. Lab. Med. 1989, 113, 381–384. [Google Scholar] [PubMed]
- Verschuur, A.C.; Vujanić, G.M.; Van Tinteren, H.; Jones, K.P.; de Kraker, J.; Sandstedt, B. Stromal and Epithelial Predominant Wilms Tumours Have an Excellent Outcome: The SIOP 93 01 Experience. Pediatr. Blood Cancer 2010, 55, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Vujanić, G.M.; Sandstedt, B. The Pathology of Wilms’ Tumour (Nephroblastoma): The International Society of Paediatric Oncology Approach. J. Clin. Pathol. 2010, 63, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, J.B.; Kiviat, N.B.; Bonadio, J.F. Nephrogenic Rests, Nephroblastomatosis, and the Pathogenesis of Wilms’ Tumor. Pediatr. Pathol. 1990, 10, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Vujanić, G.M.; Apps, J.R.; Moroz, V.; Ceroni, F.; Williams, R.D.; Sebire, N.J.; Pritchard-Jones, K. Nephrogenic Rests in Wilms Tumors Treated with Preoperative Chemotherapy: The UK SIOP Wilms Tumor 2001 Trial Experience. Pediatr. Blood Cancer 2017, 64, e26547. [Google Scholar] [CrossRef]
- Beckwith, J.B. Precursor Lesions of Wilms Tumor: Clinical and Biological Implications. Med. Pediatr. Oncol. 1993, 21, 158–168. [Google Scholar] [CrossRef]
- Schmidt, L.S.; Linehan, W.M. Genetic Predisposition to Kidney Cancer. Semin. Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Spaner, S.J.; Yu, Y.; Cook, A.J.; Boag, G. Pediatric Metanephric Adenoma: Case Report and Review of the Literature. Int. Urol. Nephrol. 2014, 46, 677–680. [Google Scholar] [CrossRef]
- Wobker, S.E.; Matoso, A.; Pratilas, C.A.; Mangray, S.; Zheng, G.; Lin, M.T.; Debeljak, M.; Epstein, J.I.; Argani, P. Metanephric Adenoma-Epithelial Wilms Tumor Overlap Lesions: An Analysis of Braf Status. Am. J. Surg. Pathol. 2019, 43, 1157–1169. [Google Scholar] [CrossRef]
- Moch, H.H.P.; Ulbright, T.M.; Reuter, V.E. World Health Organization Classification of Tumors of the Urinary System and Male Genital Organs; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Kuroda, N.; Toi, M.; Hiroi, M.; Enzan, H. Review of Chromophobe Renal Cell Carcinoma with Focus on Clinical and Pathobiological Aspects. Histol. Histopathol. 2003, 18, 165–171. [Google Scholar] [PubMed]
- Kuroda, N.; Toi, M.; Hiroi, M.; Enzan, H. Review of Papillary Renal Cell Carcinoma with Focus on Clinical and Pathobiological Aspects. Histol. Histopathol. 2003, 18, 487–494. [Google Scholar] [PubMed]
- Lopez, J.I. Renal Tumors with Clear Cells. A Review. Pathol. Res. Pract. 2013, 209, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.E.; Illei, P.B.; Allaf, M.; Gonzalez, N.; Morris, K.; Hicks, J.; Demarzo, A.; Reuter, V.E.; Amin, M.B.; Epstein, J.I.; et al. t(6;11) Renal Cell Carcinoma (RCC): Expanded Immunohistochemical Profile Emphasizing Novel RCC Markers and Report of 10 New Genetically Confirmed Cases. Am. J. Surg. Pathol. 2014, 38, 604–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorner, P.S.; Shago, M.; Marrano, P.; Shaikh, F.; Somers, G.R. TFE3-Positive Renal Cell Carcinomas Are Not Always Xp11 Translocation Carcinomas: Report of a Case with a TPM3-ALK Translocation. Pathol. Res. Pract. 2016, 212, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Roma, A.; Magi-Galluzzi, C. The Usefulness of Immunohistochemical Markers in the Differential Diagnosis of Renal Neoplasms. Clin. Lab. Med. 2005, 25, 247–257. [Google Scholar] [CrossRef]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.H.A.G.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Auvil, J.M.G.; Meerzaman, D.; Chen, Q.R.; et al. A Children’s Oncology Group and Target Initiative Exploring the Genetic Landscape of Wilms Tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.E.; Gadd, S.; Huff, V.; Gerhard, D.S.; Dome, J.S.; Perlman, E.J. A Unique Subset of Low-Risk Wilms Tumors Is Characterized by Loss of Function of TRIM28 (KAP1), a Gene Critical in Early Renal Development: A Children’s Oncology Group Study. PLoS ONE 2018, 13, e0208936. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; Vocke, C.D.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar]
- Green, W.M.; Yonescu, R.; Morsberger, L.; Morris, K.; Netto, G.J.; Epstein, J.I.; Illei, P.B.; Allaf, M.; Ladanyi, M.; Griffin, C.A.; et al. Utilization of a TFE3 Break-Apart Fish Assay in a Renal Tumor Consultation Service. Am. J. Surg. Pathol. 2013, 37, 1150–1163. [Google Scholar] [CrossRef]
- Pradhan, D.; Roy, S.; Quiroga-Garza, G.; Cieply, K.; Mahaffey, A.L.; Bastacky, S.; Dhir, R.; Parwani, A.V. Validation and Utilization of a TFE3 Break-Apart Fish Assay for Xp11.2 Translocation Renal Cell Carcinoma and Alveolar Soft Part Sarcoma. Diagn. Pathol. 2015, 10, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argani, P.; Yonescu, R.; Morsberger, L.; Morris, K.; Netto, G.J.; Smith, N.; Gonzalez, N.; Illei, P.B.; Ladanyi, M.; Griffin, C.A. Molecular Confirmation of t(6;11)(P21;Q12) Renal Cell Carcinoma in Archival Paraffin-Embedded Material Using a Break-Apart TFEb FISH Assay Expands Its Clinicopathologic Spectrum. Am. J. Surg. Pathol. 2012, 36, 1516–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Wang, Y.; Jiang, Y.; Zhang, W.; Li, Y. Genetic Analysis and Clinicopathological Features of ALK-Rearranged Renal Cell Carcinoma in a Large Series of Resected Chinese Renal Cell Carcinoma Patients and Literature Review. Histopathology 2017, 71, 53–62. [Google Scholar] [CrossRef]
- Debelenko, L.V.; Raimondi, S.C.; Daw, N.; Shivakumar, B.R.; Huang, D.; Nelson, M.; Bridge, J.A. Renal Cell Carcinoma with Novel VCL-ALK Fusion: New Representative of ALK-Associated Tumor Spectrum. Mod. Pathol. 2011, 24, 430–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoolmeester, J.K.; Cheville, J.C.; Folpe, A.L. Synovial Sarcoma of the Kidney: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 16 Cases. Am. J. Surg. Pathol. 2014, 38, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Zhao, S.; Said, J.W.; Merino, M.J.; Adeniran, A.J.; Xie, Z.; Nawaf, C.B.; Choi, J.; Belldegrun, A.S.; Pantuck, A.J.; et al. Genomic Characterization of Sarcomatoid Transformation in Clear Cell Renal Cell Carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 2170–2175. [Google Scholar] [CrossRef] [Green Version]
- Rudzinski, E.R.; Lockwood, C.M.; Stohr, B.A.; Vargas, S.O.; Sheridan, R.; Black, J.O.; Rajaram, V.; Laetsch, T.W.; Davis, J.L. Pan-Trk Immunohistochemistry Identifies NTRK Rearrangements in Pediatric Mesenchymal Tumors. Am. J. Surg. Pathol. 2018, 42, 927–935. [Google Scholar] [CrossRef]
- Marsden, L.; Jennings, L.J.; Gadd, S.; Yu, M.; Perlman, E.J.; Cajaiba, M.M. BRAF Exon 15 Mutations in Pediatric Renal Stromal Tumors: Prevalence in Metanephric Stromal Tumors. Hum. Pathol. 2017, 60, 32–36. [Google Scholar] [CrossRef]
- Yang, L.; Wang, K.; Hong, L.; Wang, Y.; Li, X. The Value of Immunohistochemistry in Diagnosing Primary Renal Synovial Sarcoma: A Case Report and Literature Review. Int. Surg. 2012, 97, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Valind, A.; Gisselsson, D. BCOR Internal Tandem Duplication and YWHAE-NUTMB2b/E Fusion Are Mutually Exclusive Events in Clear Cell Sarcoma of the Kidney. Genes Chromosom. Cancer 2016, 55, 120–123. [Google Scholar] [CrossRef]
- Kenny, C.; Bausenwein, S.; Lazaro, A.; Furtwangler, R.; Gooskens, S.L.; van den Heuvel Eibrink, M.; Vokuhl, C.; Leuschner, I.; Graf, N.; Gessler, M.; et al. Mutually Exclusive BCOR Internal Tandem Duplications and YWHAE-NUTM2 Fusions in Clear Cell Sarcoma of Kidney: Not the Full Story. J. Pathol. 2016, 238, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Wegert, J.; Vokuhl, C.; Collord, G.; Del Castillo Velasco-Herrera, M.; Farndon, S.J.; Guzzo, C.; Jorgensen, M.; Anderson, J.; Slater, O.; Duncan, C.; et al. Recurrent Intragenic Rearrangements of EGFR and BRAF in Soft Tissue Tumors of Infants. Nat. Commun. 2018, 9, 2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vokuhl, C.; Nourkami-Tutdibi, N.; Furtwangler, R.; Gessler, M.; Graf, N.; Leuschner, I. ETV6-NTRK3 in Congenital Mesoblastic Nephroma: A Report of the SIOP/GPOH Nephroblastoma Study. Pediatr. Blood Cancer 2018, 65, e26925. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Lee, J.; Netto, G.J.; Zheng, G.; Tseh-Lin, M.; Park, B.H. Frequent BRAF V600e Mutations in Metanephric Stromal Tumor. Am. J. Surg. Pathol. 2016, 40, 719–722. [Google Scholar] [CrossRef]
- Tan, Y.S.; Ng, L.G.; Yip, S.K.; Tay, M.H.; Lim, A.S.; Tien, S.L.; Cheng, L.; Tan, P.H. Synovial Sarcoma of the Kidney: A Report of 4 Cases with Pathologic Appraisal and Differential Diagnostic Review. Anal. Quant. Cytol. Histol. 2010, 32, 239–245. [Google Scholar]
- Murphy, W.M.; Grignon, D.J.; Perlman, E.J. Afip Atlas of Tumor Pathology Series 4; Tumors of the Kidney, Bladder, and Related Urinary Structures. 1; The American Registry of Pathology: Washington, DC, USA, 2004; pp. 10–28. [Google Scholar]
- Goto, S.; Umehara, S.; Gerbing, R.B.; Stram, D.O.; Brodeur, G.M.; Seeger, R.C.; Lukens, J.N.; Matthay, K.K.; Shimada, H. Histopathology (International Neuroblastoma Pathology Classification) and MYCN Status in Patients with Peripheral Neuroblastic Tumors: A Report from the Children’s Cancer Group. Cancer 2001, 92, 2699–2708. [Google Scholar] [CrossRef]
- Wang, L.L.; Perlman, E.J.; Vujanić, G.M.; Zuppan, C.; Brundler, M.A.; Cheung, C.R.; Calicchio, M.L.; Dubois, S.; Cendron, M.; Murata-Collins, J.L.; et al. Desmoplastic Small Round Cell Tumor of the Kidney in Childhood. Am. J. Surg. Pathol. 2007, 31, 576–584. [Google Scholar] [CrossRef]
- Mora, J.; Modak, S.; Cheung, N.K.; Meyers, P.; de Alava, E.; Kushner, B.; Magnan, H.; Tirado, O.M.; Laquaglia, M.; Ladanyi, M.; et al. Desmoplastic Small Round Cell Tumor 20 Years after Its Discovery. Future Oncol. 2015, 11, 1071–1081. [Google Scholar] [CrossRef]
- Murugan, P.; Rao, P.; Tamboli, P.; Czerniak, B.; Guo, C.C. Primary Ewing Sarcoma/Primitive Neuroectodermal Tumor of the Kidney: A Clinicopathologic Study of 23 Cases. Pathol. Oncol. Res. 2018, 24, 153–159. [Google Scholar] [CrossRef]
- Kao, Y.C.; Owosho, A.A.; Sung, Y.S.; Zhang, L.; Fujisawa, Y.; Lee, J.C.; Wexler, L.; Argani, P.; Swanson, D.; Dickson, B.C.; et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases with Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am. J. Surg. Pathol. 2018, 42, 604–615. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas with CIC-Rearrangements Are a Distinct Pathologic Entity with Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am. J. Surg. Pathol. 2017, 41, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Ohe, C.; Smith, S.C.; Sirohi, D.; Divatia, M.; de Peralta-Venturina, M.; Paner, G.P.; Agaimy, A.; Amin, M.B.; Argani, P.; Chen, Y.B.; et al. Reappraisal of Morphologic Differences between Renal Medullary Carcinoma, Collecting Duct Carcinoma, and Fumarate Hydratase-Deficient Renal Cell Carcinoma. Am. J. Surg. Pathol. 2018, 42, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Billis, A.; Shah, R.B.; Moch, H.; Osunkoya, A.O.; Jochum, W.; Hes, O.; Bacchi, C.E.; de Castro, M.G.; Hansel, D.E.; et al. Carcinoma of the Collecting Ducts of Bellini and Renal Medullary Carcinoma: Clinicopathologic Analysis of 52 Cases of Rare Aggressive Subtypes of Renal Cell Carcinoma with a Focus on Their Interrelationship. Am. J. Surg. Pathol. 2012, 36, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Yoon, G.; Ro, J.Y. Clinicopathologic and Molecular Pathology of Collecting Duct Carcinoma and Related Renal Cell Carcinomas. Adv. Anat. Pathol. 2017, 24, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.D.; Chagtai, T.; Alcaide-German, M.; Apps, J.; Wegert, J.; Popov, S.; Vujanić, G.; van Tinteren, H.; van den Heuvel-Eibrink, M.M.; Kool, M.; et al. Multiple Mechanisms of MYCN Dysregulation in Wilms Tumour. Oncotarget 2015, 6, 7232–7243. [Google Scholar] [CrossRef] [Green Version]
- Kalimuthu, S.N.; Chetty, R. Gene of the Month: SMARCB1. J. Clin. Pathol. 2016, 69, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Haswell, J.R.; Roberts, C.W. Molecular Pathways: SWI/SNF (BAF) Complexes Are Frequently Mutated in Cancer—Mechanisms and Potential Therapeutic Insights. Clin. Cancer Res. 2014, 20, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lee, R.S.; Alver, B.H.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.M.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-Mediated SWI/SNF Complex Function Is Essential for Enhancer Regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef]
- Sredni, S.T.; Tomita, T. Rhabdoid Tumor Predisposition Syndrome. Pediatr. Dev. Pathol. 2015, 18, 49–58. [Google Scholar] [CrossRef]
- Argani, P.; Kao, Y.C.; Zhang, L.; Bacchi, C.; Matoso, A.; Alaggio, R.; Epstein, J.I.; Antonescu, C.R. Primary Renal Sarcomas with BCOR-CCNB3 Gene Fusion: A Report of 2 Cases Showing Histologic Overlap with Clear Cell Sarcoma of Kidney, Suggesting Further Link between BCOR-Related Sarcomas of the Kidney and Soft Tissues. Am. J. Surg. Pathol. 2017, 41, 1702–1712. [Google Scholar] [CrossRef]
- Raj, P.; Khanolkar, A.; Sarin, Y.K. Metanephric Adenofibroma Masquerading as Wilms’ Tumor. APSP J. Case Rep. 2016, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.R.; Green, D.M.; Perlman, E.J.; Beckwith, J.B.; Argani, P. The Spectrum of Metanephric Adenofibroma and Related Lesions: Clinicopathologic Study of 25 Cases from the National Wilms Tumor Study Group Pathology Center. Am. J. Surg. Pathol. 2001, 25, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Mangray, S.; Breese, V.; Jackson, C.L.; Lombardo, K.; Taliano, R.; Resnick, M.; Yakirevich, E. Application of BRAF V600e Mutation Analysis for the Diagnosis of Metanephric Adenofibroma. Am. J. Surg. Pathol. 2015, 39, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA-methylation-based Classification of Central Nervous System Tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, Y.; Şenbabaoğlu, Y.; Ciriello, G.; Yang, L.; Reznik, E.; Shuch, B.; Micevic, G.; De Velasco, G.; Shinbrot, E.; et al. Multilevel Genomics-based Taxonomy of Renal Cell Carcinoma. Cell Rep. 2016, 14, 2476–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Paemel, R.; Vlug, R.; de Preter, K.; van Roy, N.; Speleman, F.; Willems, L.; Lammens, T.; Laureys, G.; Schleiermacher, G.; Tytgat, G.A.M.; et al. The Pitfalls and Promise of Liquid Biopsies for Diagnosing and Treating Solid Tumors in Children: A Review. Eur. J. Pediatr. 2020, 179, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Litjens, G.; Bandi, P.; Ehteshami Bejnordi, B.; Geessink, O.; Balkenhol, M.; Bult, P.; Halilovic, A.; Hermsen, M.; van de Loo, R.; Vogels, R.; et al. 1399 H&E stained Sentinel Lymph Node Sections of Breast Cancer Patients: The CAMYLEON Dataset. Gigascience 2018, 7, giy065. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Percentage (%) |
|---|---|
| WT | 88.0 |
| CCSK | 3.6 |
| MRTK | 3.0 |
| CMN | 3.0 |
| RCC | 1.8 |
| Others | 0.6 |
| Epithelial Pattern | Immunohistochemistry | Ref | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT1 (n) | AMACR | Pankeratin | CK7 | TFE3 | TFEB | Melanocytic markers | BRAF V600E | ||
| WT | ++ | −− | +/− | −− | −− | −− | −− | −− | [16,17,18,19,20,21,22,23,24,25] |
| NR | + | NA | + | −/+ * | NA | NA | NA | − | [17,18] |
| MA | ++ | − | +/− | −/+ * | NA | NA | NA | + | [17,18,20,21,22,25,26] |
| Xp11.2 tRCC | −− | ++ | −/+ * | − | ++ | −− | −/+ | −− | [14,24,26,27,28,29,30] |
| t(6;11) RCC | −− | + | − | − | −− | + | + | −− | [14,24,26,27,28,29,30] |
| PRCC | −− | ++ | ++ | + | −− | −− | −− | −− | [17,20,21,26,27,28] |
| Mesenchymal Pattern | Immunohistochemistry | Notes | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|
| WT1 (n) | NGFR | BCOR | Cyclin D1 | INI1 | CD34 | BCL-2 | |||
| WT | +/− | − | −/+ * | − | ++ | −/+ | + | Rhabdomyoblastic cells desmin + | [16,23,31,32,33,34,35,36,37] |
| CCSK | −− | ++ | + | ++ | ++ | −− | −/+ | [16,31,32,33,34,35,36,37,38,39] | |
| Cellular/Mixed CMN | −− | − * | − * | +/++ | ++ | −− | −− | [16,19,31,32,33,34,35,36,37,40,41] | |
| Classic CMN | −− | −− | − * | +/++ | ++ | −− | −− | [16,19,31,34,35,36,37,40,41] | |
| MST | +/− | − | − * | NA | ++ | ++ | NA | BRAFV600E +/− | [33,34,42,43] |
| Undifferentiated Pattern | Immunohistochemistry | Notes | Ref | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT1 (n) | INI1 | NGFR | Keratin | CD99 | NE | Desmin | PAX8 | NB84 | Cyclin D1 | |||
| WT | + a | ++ | - | +/− | − | −− | − | ++ | −− | − | [16,17,23,31,34,36,40,44,45,46,47,48,49,50,51] | |
| MRTK | − (c +/−) | −− | − * | +/− (d) * | +/− | NA | +/− c | −− | NA | ++ | [15,16,31,34,36,40,46,47,48,52] | |
| NB | − | ++ | + | − | −− | ++ | −− | −− | ++ | + | PHOXB2 + in >90% | [16,31,40,47,48,50,51,53] |
| DSRCT | + b | ++ | −− | + (d) | − | − | ++ (d) | −− | − | −/+ | [16,34,36,40,44,47,48,50,51] | |
| EWS | −− | ++ | − | − ** | ++ (m) | − | −− | −− | −/+ | ++ | Fli1+ in >85% | [16,36,45,47,48,49,50,51] |
| Tumor Type | Subtype | Genetic Aberrations Useful in Diagnostics | Techniques |
|---|---|---|---|
| WT | ~35% WT1, CTNNB1, WTX | NGS | |
| ~10% SIX1/2 | NGS | ||
| ~15% microRNA processing genes (DROSHA, DGCR8, DICER1) | NGS | ||
| ~ 2.5% TRIM28 mutations (up to 90% in pure epithelial WT) | NGS | ||
| ~5% TP53 mutations (Anaplastic WT) | IHC, NGS | ||
| Less common mutations: FXBW7, MYCN, BCOR, MLLT1 | NGS | ||
| CCSK | Somatic BCOR-ITD (85–100%) | IHC, FISH | |
| t(10;17) (~10%) | FISH | ||
| Rare: BCOR-CCNB3 translocation | FISH, NGS | ||
| MRTK | ~95% biallelic inactivation INI1 (SMARCB1) ~5% SMARCA4 mutations | IHC, NGS, NGS | |
| CMN | Classic | ~57% EGFR-ITD | NGS |
| Cellular | 70–80% t(12;15)(p13;q25) | FISH | |
| 12% EGFR-ITD | NGS | ||
| Few cases reported with BRAF-ID rearrangements | NGS | ||
| Mixed | t(12;15)(p13;q25) | FISH, NGS | |
| 82% EGFR-ITD | NGS | ||
| RCC | t(6;11) tRCC | Translocations involving TFEB | IHC (TFEB), FISH |
| Xp11.2 tRCC | Translocations involving TFE3 | IHC (TFE3), FISH | |
| PRCC | Type 1: 81% MET alterations | NGS | |
| Type 2: 25% CDKN2A alterations | NGS | ||
| ALK-RCC | Translocations involving ALK | FISH | |
| HLRCC-RCC | FH mutations | IHC, NGS | |
| SDH-related RCC | SDHB mutations | IHC, NGS | |
| Metanephric tumors | MA | ~90% BRAFV600E mutation | IHC, NGS |
| MST | ~65% BRAFV600E mutation | IHC, NGS | |
| MAF | ~50% BRAFV600E mutation | IHC, NGS | |
| Neuroblastoma | Mainly in high risk: MYCN amplification (18–38%) | FISH, NGS | |
| Mutations in a variety of genes (e.g., MYCN) | NGS | ||
| EWS | Translocations involving EWS | FISH | |
| DSRCT | >95% t(11;22)(p13;q11.2 or q12) | FISH |
| Panels | Immunohistochemical Stains Recommended in Panel | Additional Stains Which Can be Useful | ||
|---|---|---|---|---|
| Epithelial pattern | WT1 | CK7 | PAX8 | INI-1 |
| Pan-cytokeratin | TFE3 * | BCOR * | CD10 | |
| Melanocytic markers | TFEB * | Cathepsin K | Vimentin | |
| AMACR | BRAFV600E * | ALK | CyclinD1 | |
| 2SC/FH | ||||
| Mesenchymal pattern | WT1 | BCL-2 | INI1 | pan-Trk * |
| CD34 | NGFR * | CD99 | TLE1 * | |
| Cyclin D1 | BCOR * | BRAFV600E * | ||
| Undifferentiated pattern | WT1 | Neuroendocrine markers | CD56 | BCOR * |
| INI1 | Cyclin D1 | CD99 | PHOX2B * | |
| Keratins | NB84 * | CD45 | ||
| Desmin | NGFR * | |||
| Tumor Type | Translocation | Fusion | FISH Probe |
|---|---|---|---|
| Xp11.2 tRCC | t(X;17)(p11.2;q25) | TFE3-ASPL | TFE3 break-apart |
| t(X;1)(p11.2;q21) | TFE3-PRCC | ||
| t(X;1)(p11.2;p34) | TFE3-PSF | ||
| (X;X)(p11.2;q12) | TFE3-NonO | ||
| t(X;17)(p11.2;q23) | TFE3-CLRC | ||
| t(6;11) tRCC | t(6;11)(p21;q12) | TFEB-MALAT1 | TFEB break-apart |
| CCSK | t(10;17)(q22;p13) | YWHAE-NUTM2B/E | YWHAE or NUTM2 break-apart |
| inv(X)(p11.4;p11.22) | BCOR-CCNB3 | BCOR-CCNB3 fusion | |
| CMN | t(12;15) (p13;q25) | ETV6-NTRK3 | ETV6 or NTRK3 break-apart; ETV6-NTRK3 fusion |
| DSRCT | t(11;22)(q13;q12) | WT1-EWS | EWS break-apart |
| EWS-rearranged sarcomas | t(11;22)(q13;q12) | WT1-EWS | EWS break-apart |
| t(11;22)((q24;q12) | FLI-1-EWS | ||
| t(21;22)(q12q12) | ERG-EWS | ||
| t(7;22)(p22;q12) | ETV1-EWS | ||
| t(17;22)(q12;q12) | E1AF-EWS | ||
| t(2;22)(q33;q12) | FEV-EWS | ||
| EWS-negative sarcomas | t(4;19) or t(10;19 | CIC-DUX4 | CIC or DUX4 break-apart |
| inv(X)(p11.4;p11.22) | BCOR-CCNB3 | BCOR-CCNB3 fusion | |
| ALK-RCC | t(2;10)(p23;q22) | VCL-ALK | ALK break-apart |
| t(1;2)(q25;p23) | TPM3-ALK | ||
| t(1;2)(p32;p23) | HOOK-ALK |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ooms, A.H.A.G.; Vujanić, G.M.; D’Hooghe, E.; Collini, P.; L’Herminé-Coulomb, A.; Vokuhl, C.; Graf, N.; van den Heuvel-Eibrink, M.M.; de Krijger, R.R. Renal Tumors of Childhood—A Histopathologic Pattern-Based Diagnostic Approach. Cancers 2020, 12, 729. https://doi.org/10.3390/cancers12030729
Ooms AHAG, Vujanić GM, D’Hooghe E, Collini P, L’Herminé-Coulomb A, Vokuhl C, Graf N, van den Heuvel-Eibrink MM, de Krijger RR. Renal Tumors of Childhood—A Histopathologic Pattern-Based Diagnostic Approach. Cancers. 2020; 12(3):729. https://doi.org/10.3390/cancers12030729
Chicago/Turabian StyleOoms, Ariadne H.A.G., Gordan M. Vujanić, Ellen D’Hooghe, Paola Collini, Aurore L’Herminé-Coulomb, Christian Vokuhl, Norbert Graf, Marry M. van den Heuvel-Eibrink, and Ronald R. de Krijger. 2020. "Renal Tumors of Childhood—A Histopathologic Pattern-Based Diagnostic Approach" Cancers 12, no. 3: 729. https://doi.org/10.3390/cancers12030729
APA StyleOoms, A. H. A. G., Vujanić, G. M., D’Hooghe, E., Collini, P., L’Herminé-Coulomb, A., Vokuhl, C., Graf, N., van den Heuvel-Eibrink, M. M., & de Krijger, R. R. (2020). Renal Tumors of Childhood—A Histopathologic Pattern-Based Diagnostic Approach. Cancers, 12(3), 729. https://doi.org/10.3390/cancers12030729