MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
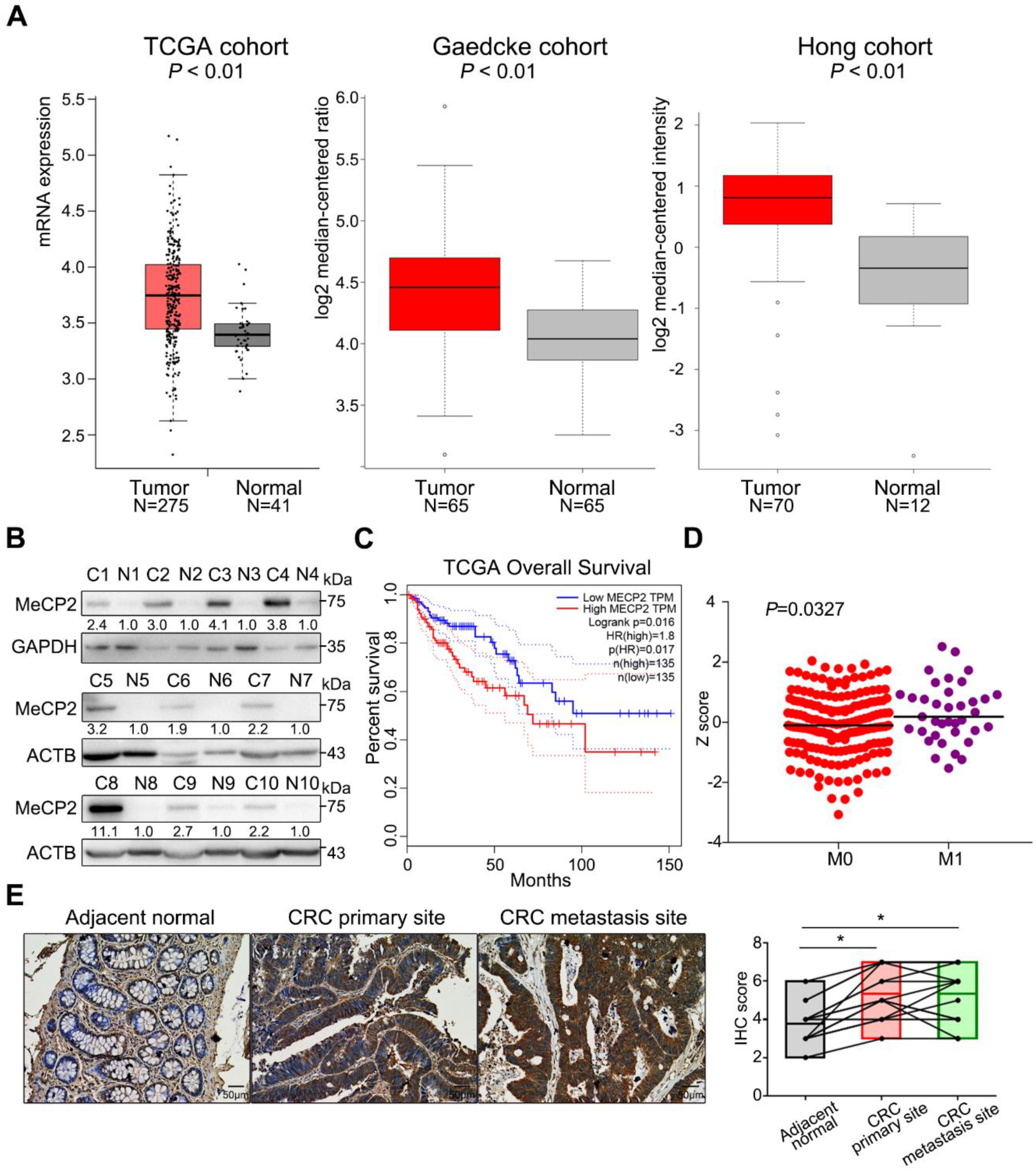
2.1. MeCP2 is Overexpressed in CRC and is Associated with A Poor Prognosis
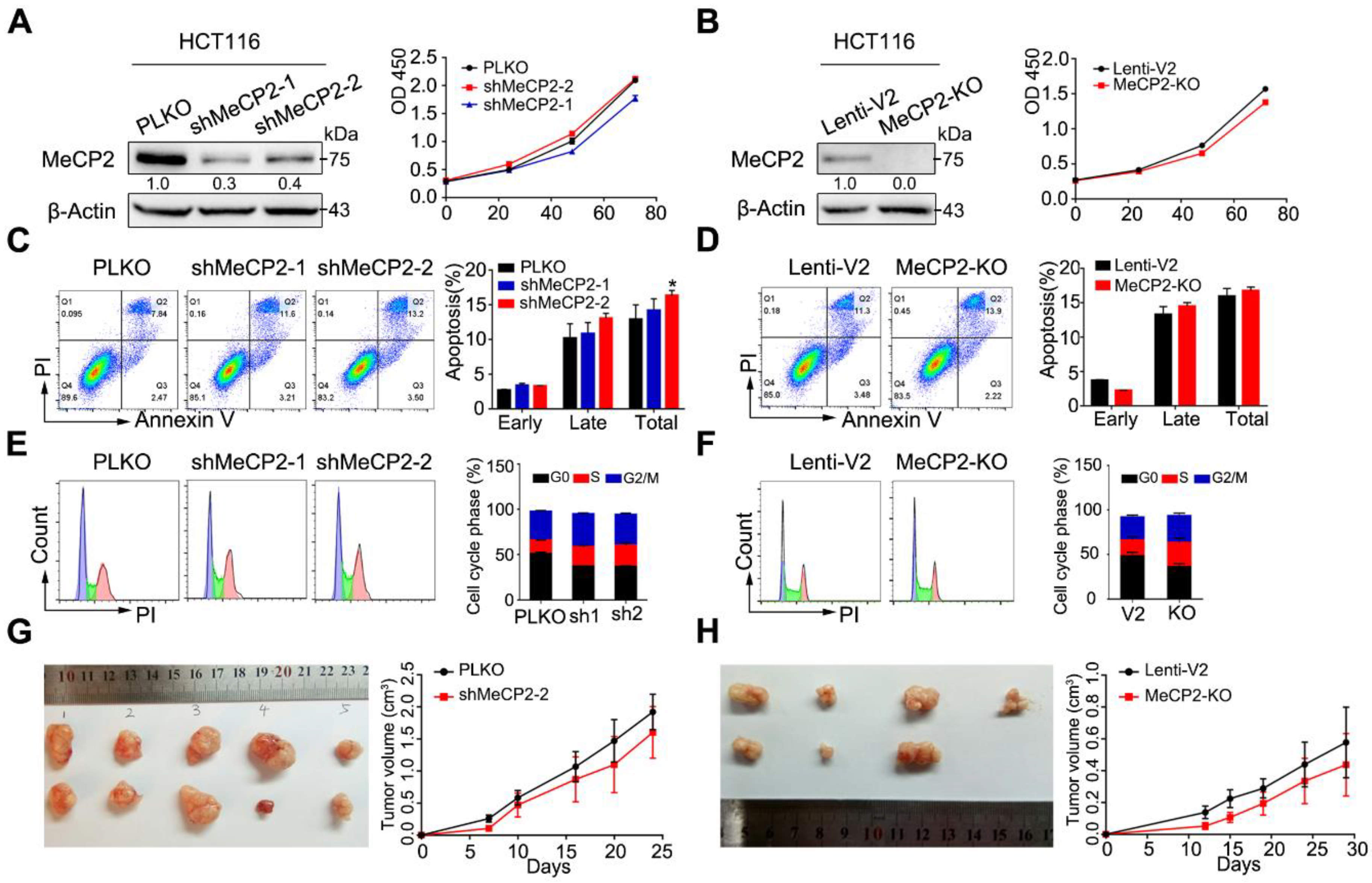
2.2. MeCP2 Depletion does not Affect the Proliferation of CRC Cells
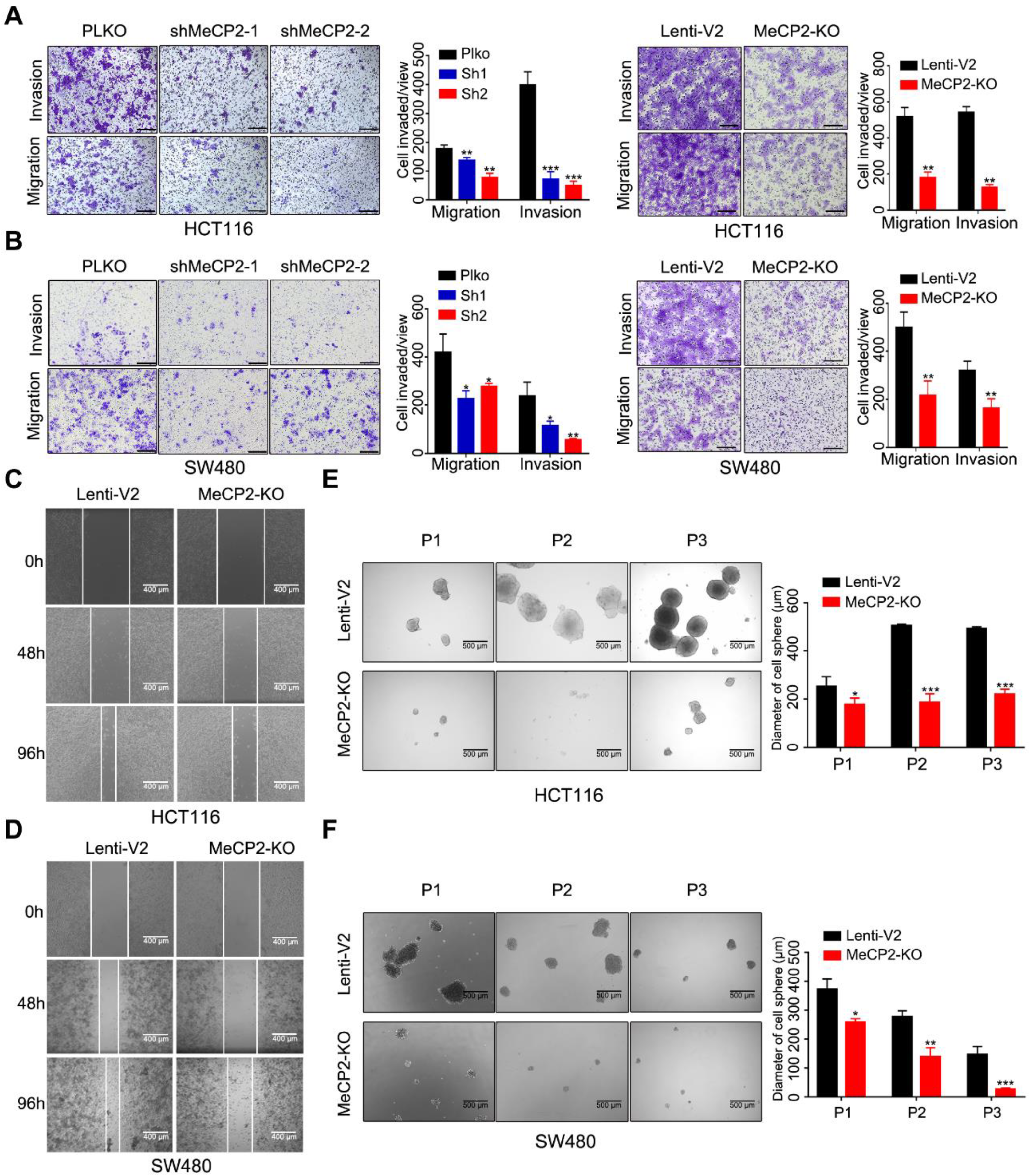
2.3. Abrogation of MeCP2 Inhibits CRC Cell Migration, Invasion and Stemness
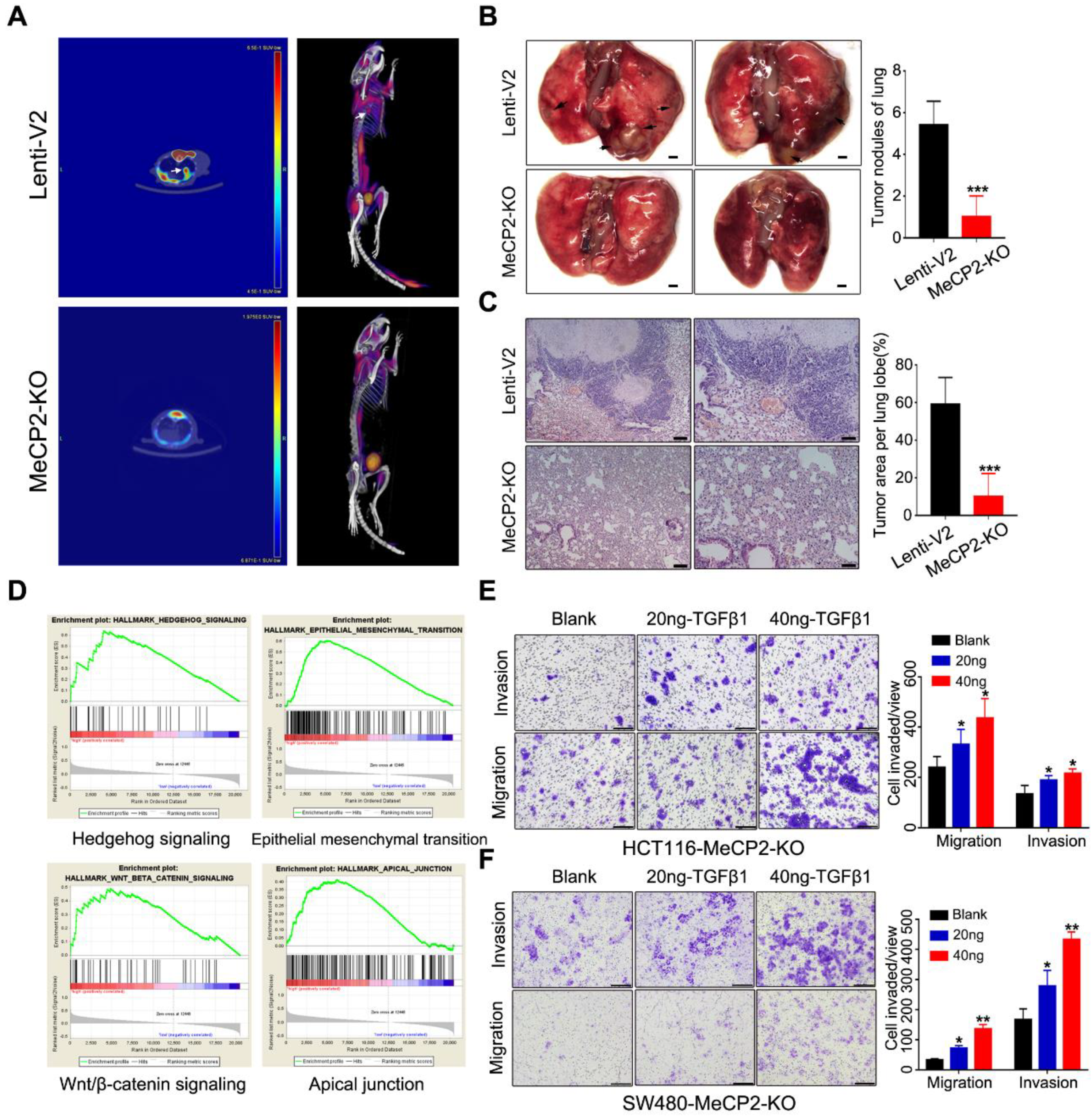
2.4. MeCP2 Knockout Suppresses Metastasis in Nude Mice
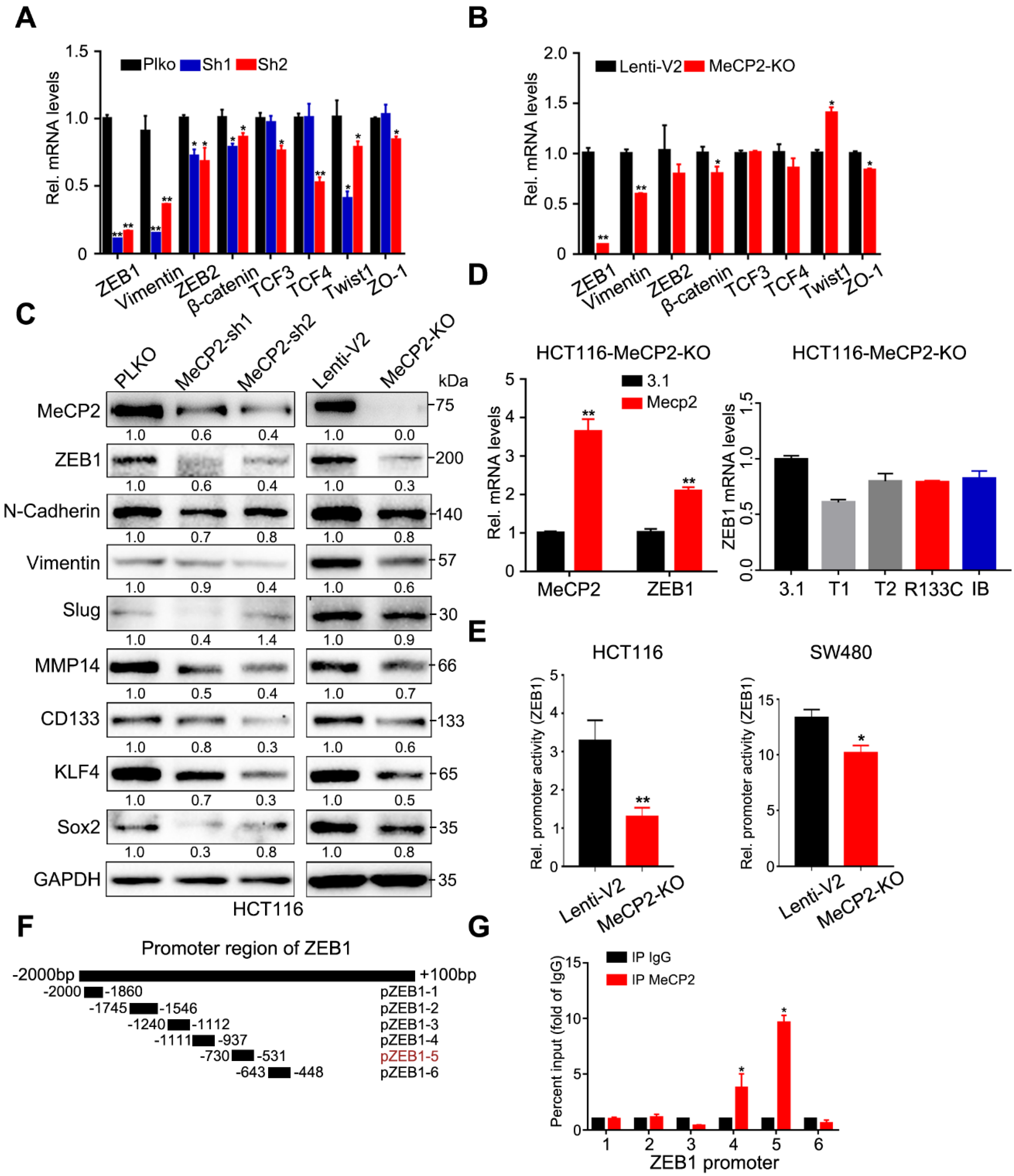
2.5. ZEB1 is the Major Effector of MeCP2
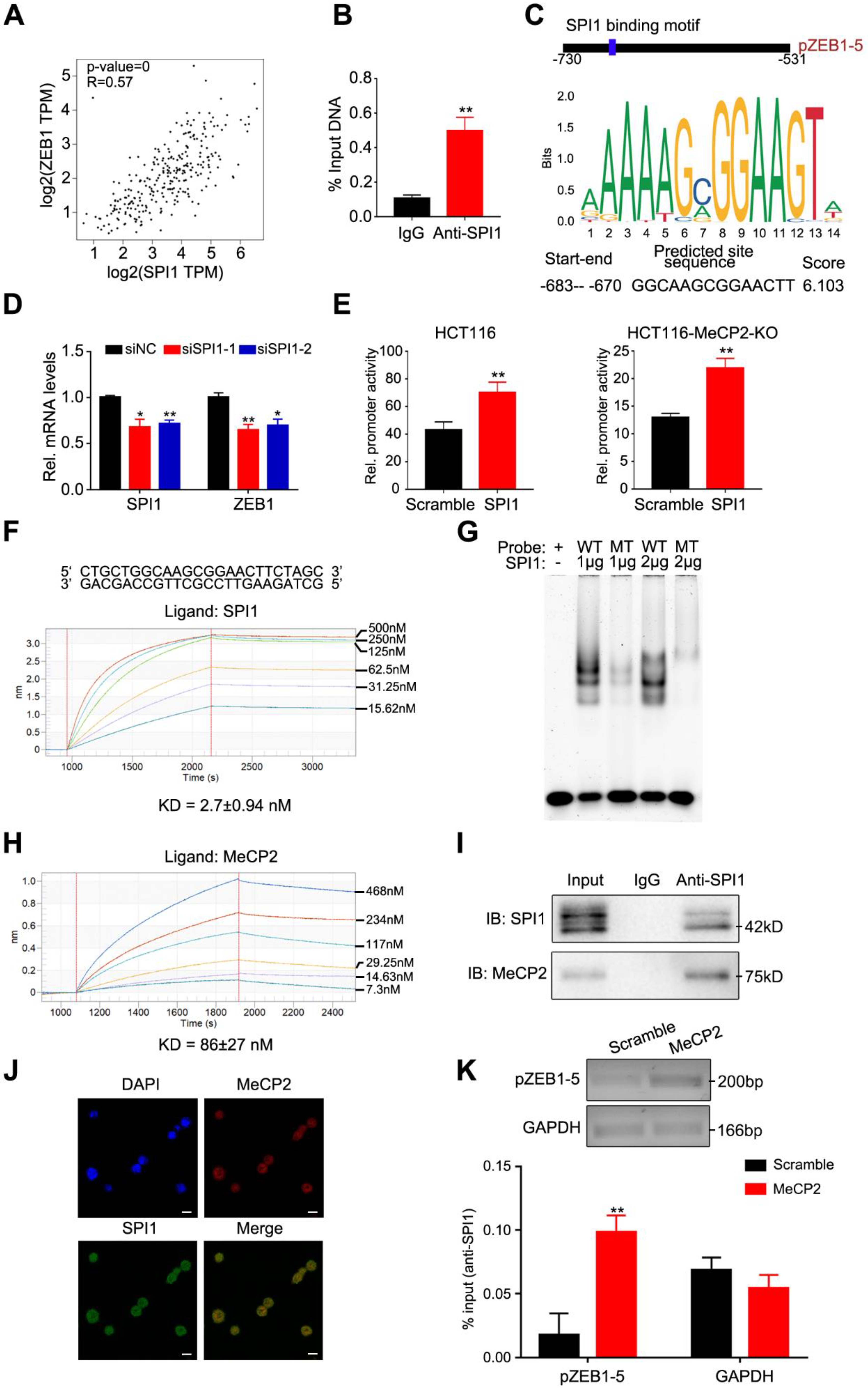
2.6. ZEB1 is A Transcriptional Target of the MeCP2-SPI1 Complex
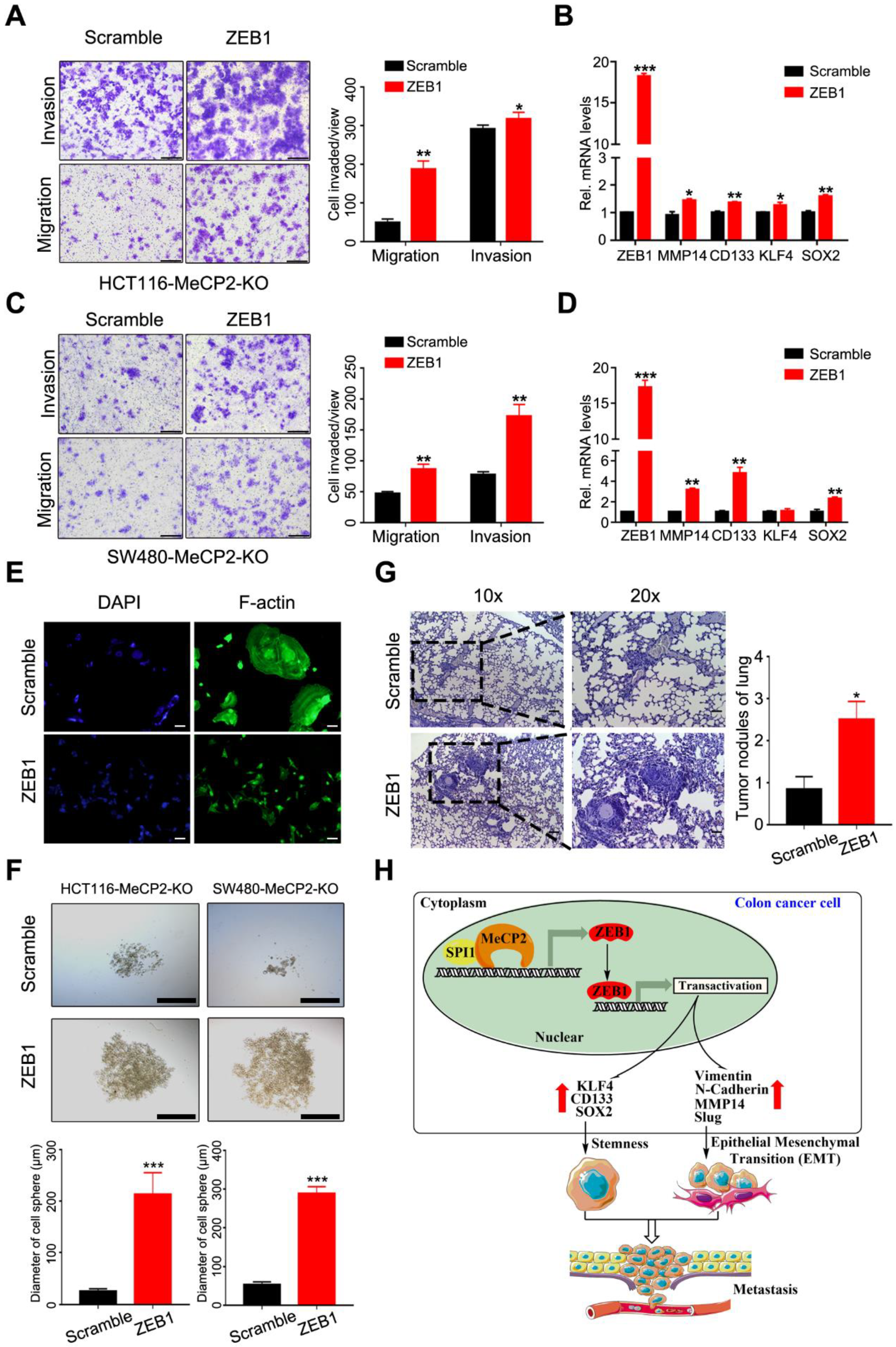
2.7. ZEB1 Restores MeCP2-Mediated Metastasis in MeCP2-Depleted CRC Cells
3. Discussion
4. Materials and Methods
4.1. Patients and Tumor Tissue Specimens
4.2. Lentiviral Infection, Establishment of Stable Cell Lines, and CRISPR Generation
4.3. Chromatin immunoprecipitation (CHIP)-qPCR
4.4. Electrophoretic Mobility Shift Assay (EMSA)
4.5. Dual Luciferase Assays
4.6. Bio-Layer Interferometry (BLI) Assays
4.7. Gene Set Enrichment Analysis (GSEA)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | colorectal cancer |
| IHC | immunohistochemistry |
| MeCP2 | Methyl-CpG binding protein 2 |
| SPI1 | Spi-1 Proto-Oncogene |
| ZEB1 | Zinc Finger E-Box Binding Homeobox 1 |
| MMP14 | Matrix Metallopeptidase 14 |
| CD133 | Prominin-Like Protein 1 |
| SOX2 | SRY-Related HMG-Box Gene 2 |
References
- Araghi, M.; Soerjomataram, I.; Jenkins, M.; Brierley, J.; Morris, E.; Bray, F.; Arnold, M. Global trends in colorectal cancer mortality: Projections to the year 2035. Int. J. Cancer 2019, 144, 2992–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hiller, J.G.; Perry, N.J.; Poulogiannis, G.; Riedel, B.; Sloan, E.K. Perioperative events influence cancer recurrence risk after surgery. Nat. Rev. Clin. Oncol. 2018, 15, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, R.I.; Smith, B.O.; Nan, X.; Free, A.; Soteriou, A.; Uhrin, D.; Bird, A.P.; Barlow, P.N. The solution structure of the domain from MeCP2 that binds to methylated DNA. J. Mol. Biol. 1999, 291, 1055–1065. [Google Scholar] [CrossRef]
- Neupane, M.; Clark, A.P.; Landini, S.; Birkbak, N.J.; Eklund, A.C.; Lim, E.; Culhane, A.C.; Barry, W.T.; Schumacher, S.E.; Beroukhim, R.; et al. MECP2 Is a Frequently Amplified Oncogene with a Novel Epigenetic Mechanism That Mimics the Role of Activated RAS in Malignancy. Cancer Discov. 2016, 6, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Hite, K.C.; Adams, V.H.; Hansen, J.C. Recent advances in MeCP2 structure and functionThis paper is one of a selection of papers published in this Special Issue, entitled 29th Annual International Asilomar Chromatin and Chromosomes Conference, and has undergone the Journal’s usual peer review process. Biochem. Cell Boil. 2009, 87, 219–227. [Google Scholar]
- Marian, M.; Pinar, A.; Scott, D.; Skirmantas, K.; Nathaniel, H.J.C. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Ming, G.-L.; Song, H. By hook or by crook: Multifaceted DNA-binding properties of MeCP2. Cell 2013, 152, 940–942. [Google Scholar] [CrossRef] [Green Version]
- Gabel, H.W.; Kinde, B.Z.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.E.; Veyver, I.B.V.D.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Peters, S.U.; Tavyev, Y.J.; Zhang, F.; Carvalho, C.M.; Schaaf, C.P.; Richman, R.; Fang, P.; Glaze, D.G.; Lupski, J.R.; et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 2009, 66, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.Y.; Zhang, J.; Guo, B.; Yang, J.; Han, J.; Zhao, X.G.; Wang, X.F.; Liu, L.Y.; Li, Z.F.; Song, T.S.J.C.; et al. MECP2 promotes cell proliferation by activating ERK1/2 and inhibiting p38 activity in human hepatocellular carcinoma HEPG2 cells. Cell. Mol. Biol. 2013, 59, 1876–1881. [Google Scholar]
- Tada, Y.; Brena, R.M.; Hackanson, B.; Morrison, C.; Otterson, G.A.; Plass, C. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J. Natl. Cancer Inst. 2006, 98, 396–406. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pander, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. WspÓŁczesna Onkol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, R.; Zhu, W.; Chu, H.; Yu, H.; Wei, P.; Wu, X.; Zhu, H.; Gao, H.; Liang, J.; et al. UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature 2019, 571, 127–131. [Google Scholar] [CrossRef]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Tsunekuni, K.; Konno, M.; Haraguchi, N.; Koseki, J.; Asai, A.; Matsuoka, K.; Kobunai, T.; Takechi, T.; Doki, Y.; Mori, M.; et al. CD44/CD133-Positive Colorectal Cancer Stem Cells are Sensitive to Trifluridine Exposure. Sci. Rep. 2019, 9, 14861. [Google Scholar] [CrossRef] [PubMed]
- Ishida, R.; Koyanagi-Aoi, M.; Oshima, N.; Kakeji, Y.; Aoi, T. The Tissue-Reconstructing Ability of Colon CSCs Is Enhanced by FK506 and Suppressed by GSK3 Inhibition. Mol. Cancer Res. 2017, 15, 1455–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Mizushima, T.; Yokoyama, Y.; Hirose, H.; Wu, X.; Qian, Y.; Ikehata, K.; Miyoshi, N.; Takahashi, H.; Haraguchi, N.; et al. Sox2 is associated with cancer stem-like properties in colorectal cancer. Sci. Rep. 2018, 8, 17639. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Pruitt, K. Functional assessment of MeCP2 in Rett syndrome and cancers of breast, colon, and prostate. Biochem. Cell Biol. 2017, 95, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Ausio, J. Role of MeCP2 in neurological disorders: Current status and future perspectives. Epigenomics 2018, 10, 5–8. [Google Scholar] [CrossRef]
- Nan, X.; Meehan, R.R.; Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef] [Green Version]
- Lyst, M.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; Alves, F.D.L.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Wang, Y.; Zhang, G.; Chen, H.; Dowdy, S.C.; Xiong, Y.; Liu, F.; Zhang, R.; Li, J.; Jiang, S.W. Chromatin composition alterations and the critical role of MeCP2 for epigenetic silencing of progesterone receptor-B gene in endometrial cancers. Cell. Mol. Life Sci. 2014, 71, 3393–3408. [Google Scholar] [CrossRef]
- Ramachandran, K.; Gopisetty, G.; Gordian, E.; Navarro, L.; Hader, C.; Reis, I.M.; Schulz, W.A.; Singal, R. Methylation-mediated repression of GADD45alpha in prostate cancer and its role as a potential therapeutic target. Cancer Res. 2009, 69, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Liu, Y.; Tong, D.; Qin, Y.; Yang, J.; Xue, M.; Du, N.; Liu, L.; Guo, B.; Hou, N.; et al. MeCP2 Promotes Gastric Cancer Progression Through Regulating FOXF1/Wnt5a/beta-Catenin and MYOD1/Caspase-3 Signaling Pathways. EBioMedicine 2017, 16, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A.J.S. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Bian, E.; Chen, X.; Xu, Y.; Ji, X.; Cheng, M.; Wang, H.; Fang, Z.; Zhao, B. A central role for MeCP2 in the epigenetic repression of miR-200c during epithelial-to-mesenchymal transition of glioma. J. Exp. Clin. Cancer Res. 2019, 38, 366. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Lewis, J.D.; Bird, A.P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992, 20, 5085–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, D.; Ge, W. MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription. Cancers 2020, 12, 758. https://doi.org/10.3390/cancers12030758
Luo D, Ge W. MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription. Cancers. 2020; 12(3):758. https://doi.org/10.3390/cancers12030758
Chicago/Turabian StyleLuo, Dan, and Wei Ge. 2020. "MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription" Cancers 12, no. 3: 758. https://doi.org/10.3390/cancers12030758
APA StyleLuo, D., & Ge, W. (2020). MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription. Cancers, 12(3), 758. https://doi.org/10.3390/cancers12030758