Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas and Paired Normal Mucosae Reveals Cyclin D1 Deregulation and Compensatory Effect of Cyclin D2

 ,
, 
 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
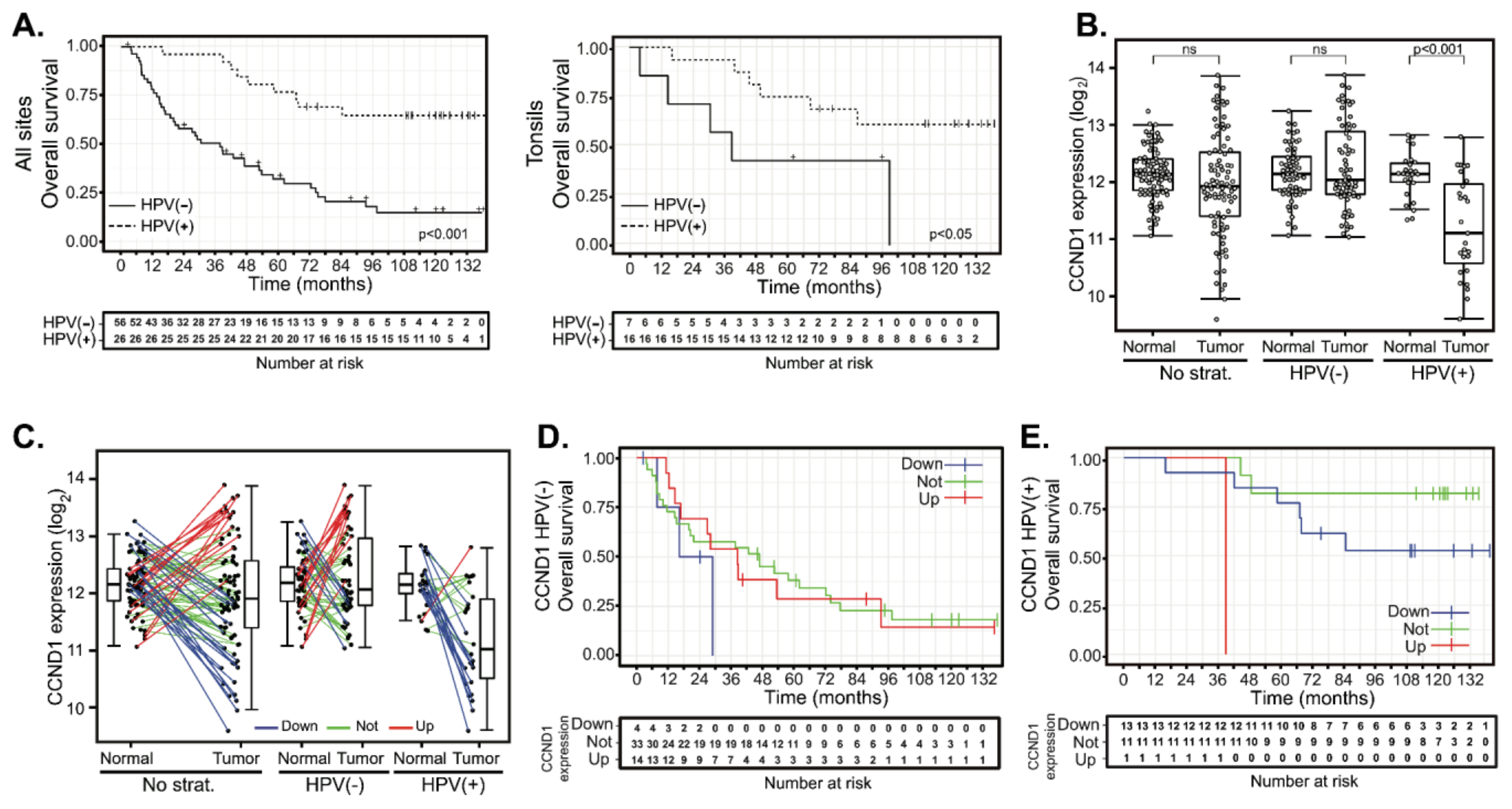
2.2. Deregulation of Cyclin D1 Expression in HPV(−) and in HPV(+) HNSCC Tumors Does Not Correlate with Patient Outcomes
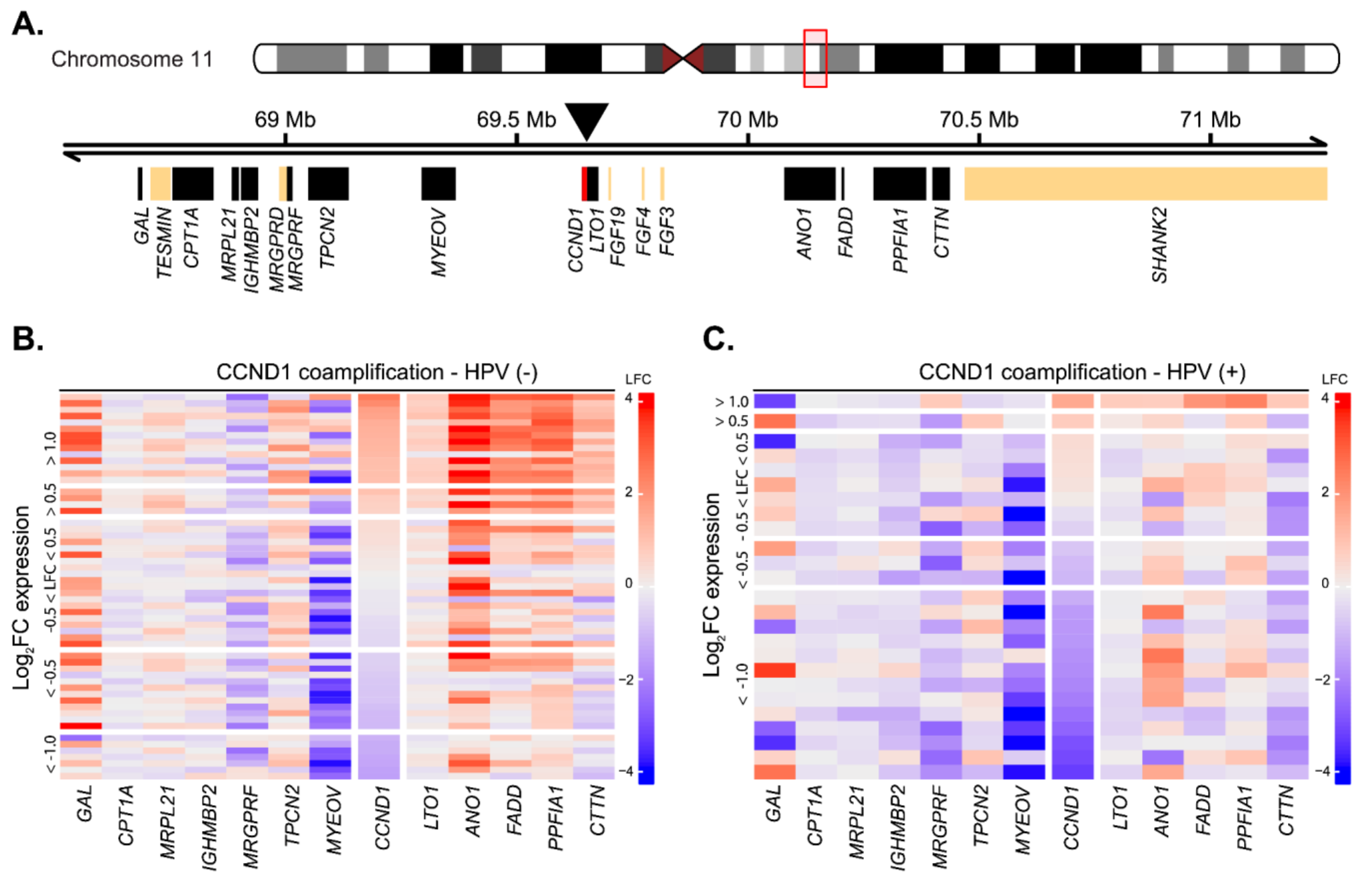
2.3. Cyclin D1 Upregulation Correlates with Bona Fide Amplification of Its Genomic Locus
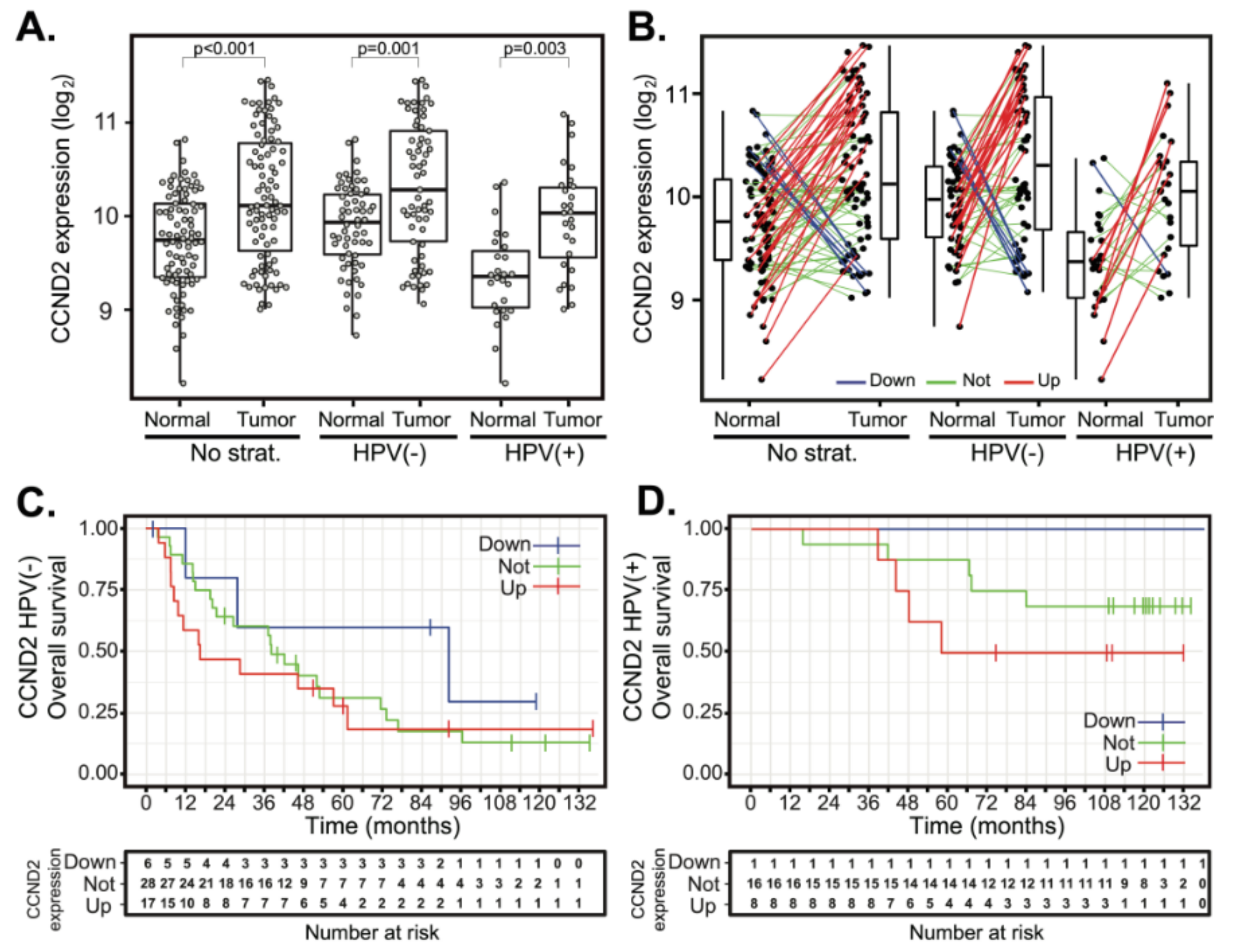
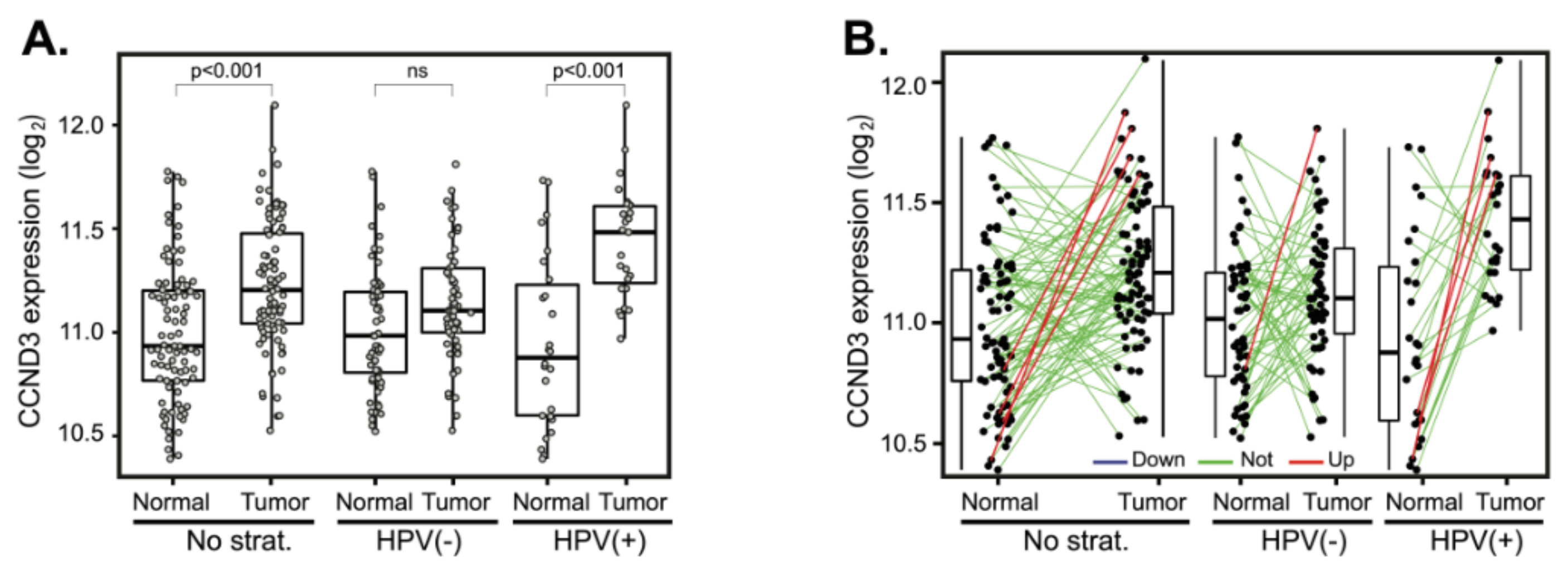
2.4. Deregulation of Cyclin D2 Does Not Correlate with Patient Outcomes
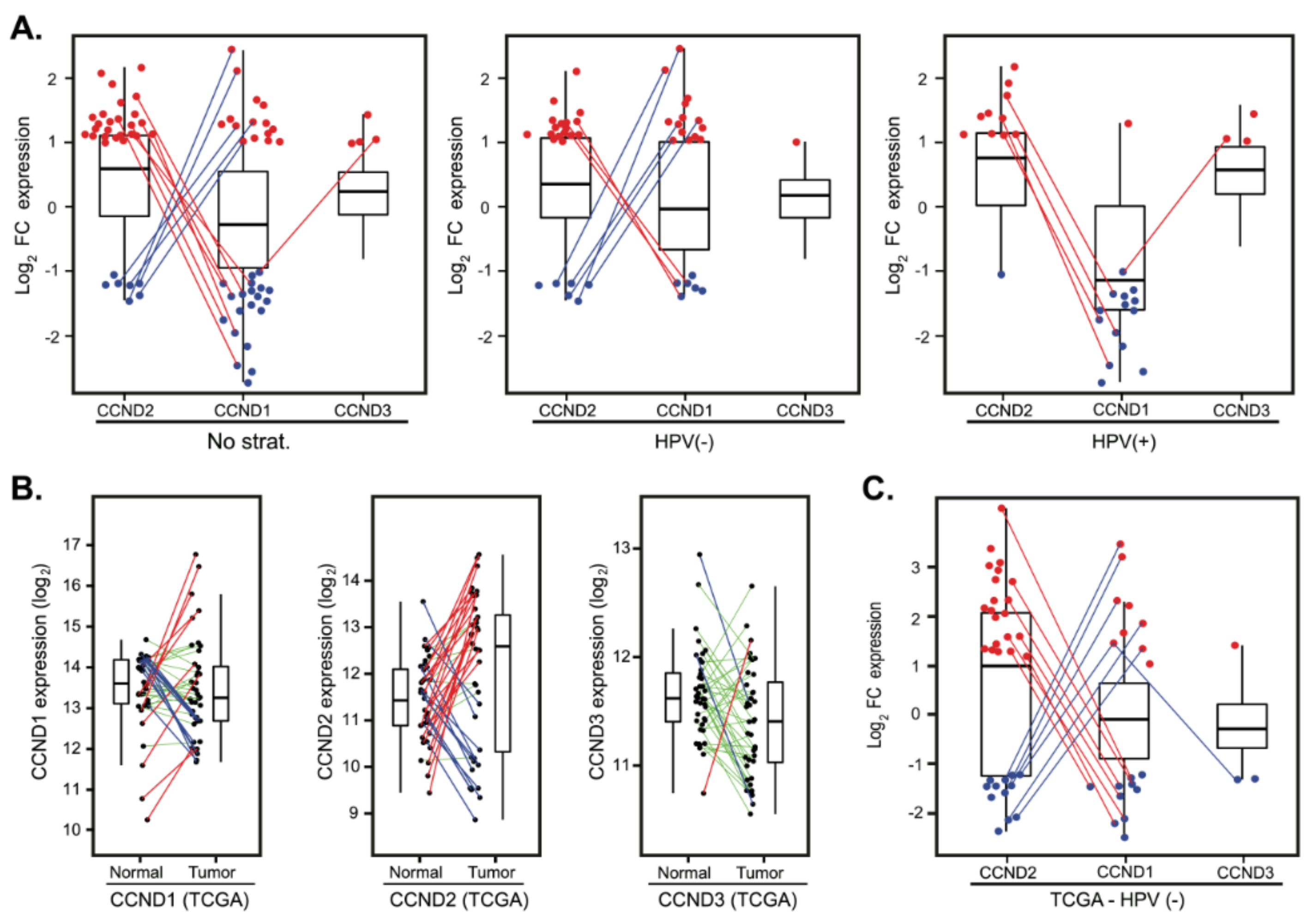
2.5. Cyclin D2 is Often Upregulated in Tumors with Downregulated Cyclin D1 and Vice Versa
2.6. Validation in an Independent Cohort
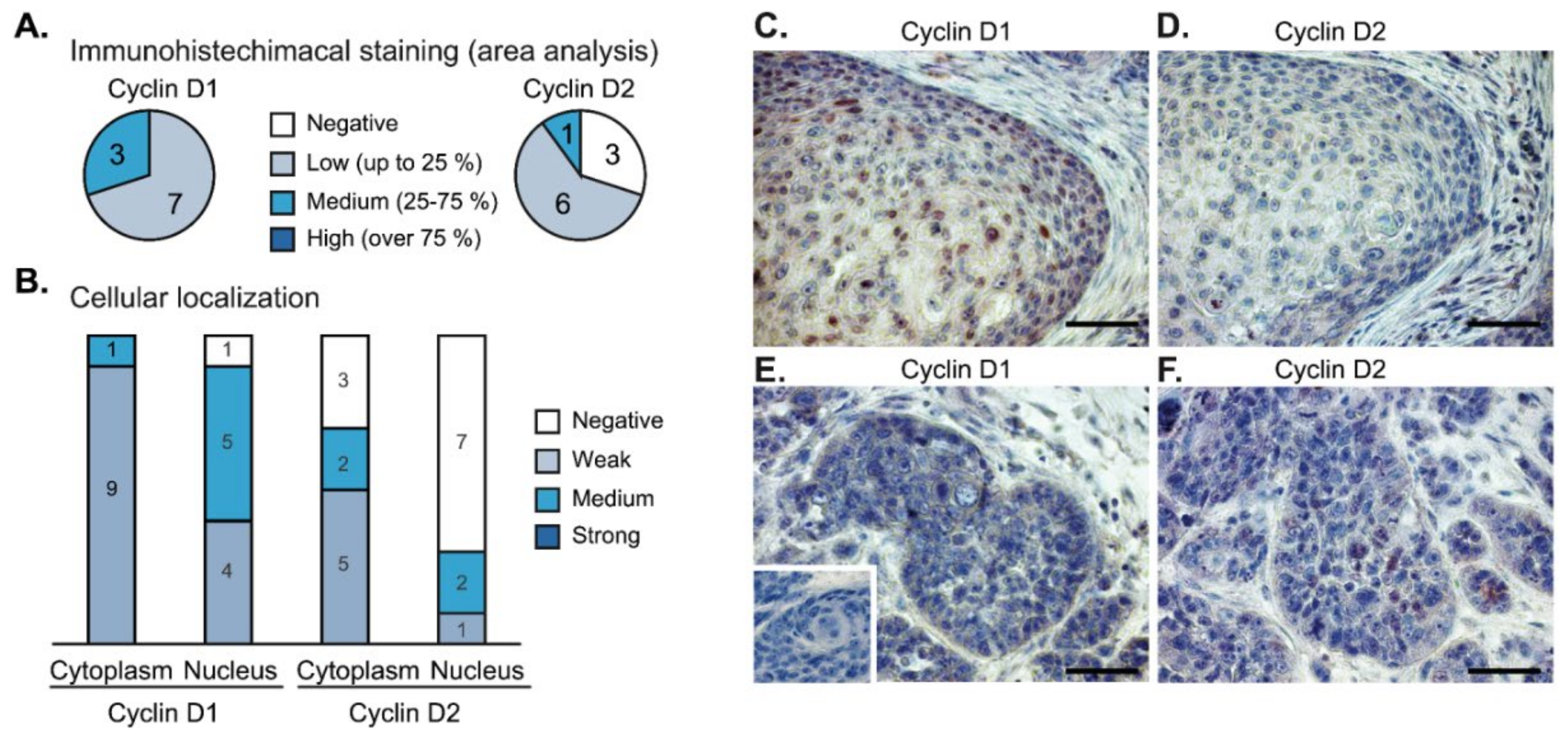
2.7. Immunohistochemical Analysis of Tumor Samples
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Transcription Profiling
4.3. Clinical Data
4.4. Analysis of CCND1 Coamplification
4.5. The Cancer Genome Atlas (TCGA) Validation Dataset
4.6. Survival Analysis
4.7. Data Availability
4.8. Ethics Approval and Consent to Participate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalavrezos, N.; Bhandari, R. Current trends and future perspectives in the surgical management of oral cancer. Oral Oncol. 2010, 46, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Grandis, J.R. Emerging drugs to treat squamous cell carcinomas of the head and neck. Expert Opin. Emerg. Drugs 2010, 15, 355–373. [Google Scholar] [CrossRef] [Green Version]
- Hashibe, M.; Brennan, P.; Chuang, S.C.; Boccia, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; et al. Interaction between Tobacco and Alcohol Use and the Risk of Head and Neck Cancer: Pooled Analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomark. Prev. 2009, 18, 541–550. [Google Scholar]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a Causal Association Between Human Papillomavirus and a Subset of Head and Neck Cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar]
- Tommasino, M. The human papillomavirus family and its role in carcinogenesis. Semin. Cancer Biol. 2014, 26, 13–21. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systemic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Psyrri, A.; Rampias, T.; Vermorken, J.B. The current and future impact of human papillomavirus on treatment of squamous cell carcinoma of the head and neck. Ann. Oncol. 2014, 25, 2101–2115. [Google Scholar] [CrossRef]
- Dayyani, F.; Etzel, C.J.; Liu, M.; Ho, C.-H.; Lippman, S.M.; Tsao, A.S. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol. 2010, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragin, C.C.R.; Modugno, F.; Gollin, S.M. The epidemiology and risk factors of head and neck cancer: A focus on human papillomavirus. J. Dent. Res. 2007, 86, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Chen, W.S.; Bindra, R.S.; Mo, A.; Hayman, T.; Husain, Z.; Contessa, J.N.; Gaffney, S.G.; Townsend, J.P.; Yu, J.B. CDKN2A Copy Number Loss Is an Independent Prognostic Factor in HPV-Negative Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2018, 8, 95. [Google Scholar] [CrossRef] [Green Version]
- Dok, R.; Nuyts, S. HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers 2016, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [Green Version]
- Bova, R.J.; Quinn, D.I.; Nankervis, J.S.; Cole, I.E.; Sheridan, B.F.; Jensen, M.J.; Morgan, G.J.; Hughes, C.J.; Sutherland, R.L. Cyclin D1 and p16INK4A expression predict reduced survival in carcinoma of the anterior tongue. Clin. Cancer Res. 1999, 5, 2810–2819. [Google Scholar] [PubMed]
- Hermida-Prado, F.; Menéndez, S.; Albornoz-Afanasiev, P.; Granda-Diaz, R.; Álvarez-Teijeiro, S.; Villaronga, M.; Allonca, E.; Alonso-Durán, L.; León, X.; Alemany, L.; et al. Distinctive Expression and Amplification of Genes at 11q13 in Relation to HPV Status with Impact on Survival in Head and Neck Cancer Patients. J. Clin. Med. 2018, 7, 501. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wawrose, J.S.; Gooding, W.E.; Garraway, L.A.; Lui, V.W.Y.; Peyser, N.D.; Grandis, J.R. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: A rational approach to preclinical model selection. Mol. Cancer Res. 2014, 12, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalides, R.; Van Veelen, N.; Hart, A.; Loftus, B.; Wientjens, E.; Balm, A. Overexpression of Cyclin D1 Correlates with Recurrence in a Group of Forty-seven Operable Squamous Cell Carcinomas of the Head and Neck. Cancer Res. 1995, 55, 975–978. [Google Scholar] [PubMed]
- Mineta, H.; Miura, K.; Takebayashi, S.; Ueda, Y.; Misawa, K.; Harada, H.; Wennerberg, J.; Dictor, M. Cyclin D1 overexpression correlates with poor prognosis in patients with tongue squamous cell carcinoma. Oral Oncol. 2000, 36, 194–198. [Google Scholar] [CrossRef]
- Wong, R.J.; Keel, S.B.; Glynn, R.J.; Varvares, M.A. Histological pattern of mandibular invasion by oral squamous cell carcinoma. Laryngoscope 2000, 110, 65–72. [Google Scholar] [CrossRef]
- Vielba, R.; Bilbao, J.; Ispizua, A.; Zabalza, I.; Alfaro, J.; Rezola, R.; Moreno, E.; Elorriaga, J.; Alonso, I.; Baroja, A.; et al. p53 and cyclin D1 as prognostic factors in squamous cell carcinoma of the larynx. Laryngoscope 2003, 113, 167–172. [Google Scholar] [CrossRef]
- Thomas, G.R.; Nadiminti, H.; Regalado, J. Molecular predictors of clinical outcome in patients with head and neck squamous cell carcinoma. Int. J. Exp. Pathol. 2005, 86, 347–363. [Google Scholar] [CrossRef]
- Rodrigo, J.P.; García-Carracedo, D.; García, L.A.; Menéndez, S.T.; Allonca, E.; González, M.V.; Fresno, M.F.; Suárez, C.; García-Pedrero, J.M. Distinctive clinicopathological associations of amplification of the cortactin gene at 11q13 in head and neck squamous cell carcinomas. J. Pathol. 2009, 217, 516–523. [Google Scholar] [CrossRef]
- Mellin, H.; Friesland, S.; Lewensohn, R.; Dalianis, T.; Munck-Wikland, E. Human papilloma virus (HPV) DNA in tonsillar cancer: Clinical correlates, risk of relapse, and survival. Int. J. Cancer 2000, 89, 300–304. [Google Scholar] [CrossRef]
- Strome, S.E.; Savva, A.; Brissett, A.E.; Gostout, B.S.; Lewis, J.; Clayton, A.C.; McGovern, R.; Weaver, A.L.; Persing, D.; Kasperbauer, J.L. Squamous cell carcinoma of the tonsils: A molecular analysis of HPV associations. Clin. Cancer Res. 2002, 8, 1093–1100. [Google Scholar]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaederlé, M.; Daniels, G.A.; Piccioni, D.E.; Fanta, P.T.; Schwab, R.B.; Shimabukuro, K.A.; Parker, B.A.; Kurzrock, R. Cyclin alterations in diverse cancers: Outcome and co-amplification network. Oncotarget 2015, 6, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossen, D.; Verhagen, C.; Van der Heijden, M.; Essers, P.; Bartelink, H.; Verheij, M.; Wessels, L.; Van den Brekel, M.; Vens, C. Genetic Factors Associated with a Poor Outcome in Head and Neck Cancer Patients Receiving Definitive Chemoradiotherapy. Cancers 2019, 11, 445. [Google Scholar] [CrossRef] [Green Version]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J.W.G.; Imoto, I.; Inoue, J.; Shimada, Y.; Ueda, M.; Imamura, M.; Bartram, C.R.; Inazawa, J. MYEOV, a gene at 11q13, is coamplified with CCND1, but epigenetically inactivated in a subset of esophageal squamous cell carcinomas. J. Hum. Genet. 2002, 47, 460–464. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Faraji, F.; Schubert, A.D.; Kagohara, L.T.; Tan, M.; Xu, Y.; Zaidi, M.; Fortin, J.-P.; Fakhry, C.; Izumchenko, E.; Gaykalova, D.A.; et al. The Genome-Wide Molecular Landscape of HPV-Driven and HPV-Negative Head and Neck Squamous Cell Carcinoma. In Molecular Determinants of Head and Neck Cancer; Humana Press: Totowa, NJ, USA, 2018; pp. 293–325. [Google Scholar] [CrossRef]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Li, X.; Hydbring, P.; Sanda, T.; Stefano, J.; Christie, A.L.; Signoretti, S.; Look, A.T.; Kung, A.L.; Von Boehmer, H.; et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012, 22, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, K.; Michikawa, Y.; Ishikawa, K.; Shoji, Y.; Iwakawa, M.; Shibahara, T.; Imai, T. Combination effects of distinct cores in 11q13 amplification region on cervical lymph node metastasis of oral squamous cell carcinoma. Int. J. Oncol. 2011, 761–769. [Google Scholar]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, E.R.; Nesbit, M.; Litwin, S.; Klein-Szanto, A.J.; Cheffetz, S.; Herlyn, M. Antisense cyclin D1 induces apoptosis and tumor shrinkage in human squamous carcinomas. Cancer Res. 1999, 59, 4876–4881. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [Green Version]
- Ciemerych, M.A.; Kenney, A.M.; Sicinska, E.; Kalaszczynska, I.; Bronson, R.T.; Rowitch, D.H.; Gardner, H.; Sicinski, P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002, 16, 3277–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Pollard, J.W. Cyclin D2 compensates for the loss of cyclin D1 in estrogen-induced mouse uterine epithelial cell proliferation. Mol. Endocrinol. 2003, 17, 1368–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, E.W.-F.; Glassford, J.; Banerji, L.; Thomas, N.S.B.; Sicinski, P.; Klaus, G.G.B. Cyclin D3 Compensates for Loss of Cyclin D2 in Mouse B-lymphocytes Activated via the Antigen Receptor and CD40. J. Biol. Chem. 2000, 275, 3479–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kempen, P.M.W.; Noorlag, R.; Braunius, W.W.; Moelans, C.B.; Rifi, W.; Savola, S.; Koole, R.; Grolman, W.; Van Es, R.J.J.; Willems, S.M. Clinical relevance of copy number profiling in oral and oropharyngeal squamous cell carcinoma. Cancer Med. 2015, 4, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.S.; Brown, B.; Whigham, A.S.; Kochaishvili, A.; Yarbrough, W.G.; Weaver, A.M. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene 2009, 28, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Arao, T.; Kato, H.; Matsumoto, K.; Terashima, M.; Hayashi, H.; De Velasco, M.A.; Fujita, Y.; Kimura, H.; Yasuda, T.; et al. Frequent amplification of oraov1 gene in esophageal squamous cell cancer promotes an aggressive phenotype via proline metabolism and ros production. Oncotarget 2014, 5, 2962–2973. [Google Scholar] [CrossRef] [Green Version]
- Al Moustafa, A.-E.; Foulkes, W.D.; Wong, A.; Jallal, H.; Batist, G.; Yu, Q.; Herlyn, M.; Sicinski, P.; Alaoui-Jamali, M.A. Cyclin D1 is essential for neoplastic transformation induced by both E6/E7 and E6/E7/ErbB-2 cooperation in normal cells. Oncogene 2004, 23, 5252–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzinger, D.; Flechtenmacher, C.; Henfling, N.; Kaden, I.; Grabe, N.; Lahrmann, B.; Schmitt, M.; Hess, J.; Pawlita, M.; Bosch, F.X. Identification of oropharyngeal squamous cell carcinomas with active HPV16 involvement by immunohistochemical analysis of the retinoblastoma protein pathway. Int. J. Cancer 2013, 133, 1389–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plath, M.; Broglie, M.A.; Förbs, D.; Stoeckli, S.J.; Jochum, W. Prognostic significance of cell cycle-associated proteins p16, pRB, cyclin D1 and p53 in resected oropharyngeal carcinoma. J. Otolaryngol. Head Neck Surg. 2018, 47, 1–9. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2014, 33, 1890–1903. [Google Scholar] [CrossRef] [Green Version]
- Michel, L.; Ley, J.; Wildes, T.M.; Schaffer, A.; Robinson, A.; Chun, S.-E.; Lee, W.; Lewis, J.; Trinkaus, K.; Adkins, D. Phase I trial of palbociclib, a selective cyclin dependent kinase 4/6 inhibitor, in combination with cetuximab in patients with recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 58, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K.; et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: A multicentre, multigroup, phase 2 trial. Lancet. Oncol. 2019, 20, 1295–1305. [Google Scholar] [CrossRef]
- Valach, J.; Fík, Z.; Strnad, H.; Chovanec, M.; Plzák, J.; Čada, Z.; Szabo, P.; Šáchová, J.; Hroudová, M.; Urbanová, M.; et al. Smooth muscle actin-expressing stromal fibroblasts in head and neck squamous cell carcinoma: Increased expression of galectin-1 and induction of poor prognosis factors. Int. J. Cancer 2012, 131, 2499–2508. [Google Scholar] [CrossRef]
- Szabó, P.; Kolář, M.; Dvořánková, B.; Lacina, L.; Štork, J.; Vlček, Č.; Strnad, H.; Tvrdek, M.; Smetana, K. Mouse 3T3 fibroblasts under the influence of fibroblasts isolated from stroma of human basal cell carcinoma acquire properties of multipotent stem cells. Biol. Cell 2011, 103, 233–248. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. Available online: http://www.r-project.org/ (accessed on 22 March 2020).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Storey, J.D.; Zhang, Y.; Torres, L.C. sva: Surrogate Variable Analysis. R package version 3.34.0. Available online: http://bioconductor.org/packages/release/bioc/html/sva.html (accessed on 22 March 2020).
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. In Statistical Genomics; Mathé, E., Davis, S., Eds.; Humana Press: New York, NY, USA, 2016; Volume 1418, pp. 391–416. ISBN 9780849331664. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2014, 6, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T. A Package for Survival Analysis in S. version 2.38. Available online: https://github.com/therneau/survival (accessed on 22 March 2020).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Scheipl, F. Survminer: Drawing Survival Curves using “ggplot2.”. Available online: https://rpkgs.datanovia.com/survminer/index.html (accessed on 22 March 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable (Stat. Signif.) | All Patients N (%) | HPV(−) N (%) | HPV(+) N (%) |
|---|---|---|---|
| No. patients | 94 | - | - |
| Sample groups | |||
| No. samples per normal mucosa | 86 | - | - |
| No. samples per tumor | 90 | 64 (71r) | 26 (29r) |
| Patients | |||
| No. patients with paired samples | 82 | 57 (70r) | 25 (30r) |
| No. patients with paired samples and known follow-up | 76 | 51 (68r) | 25 (32r) |
| Age at surgery (n.s.) | |||
| Median (range) | 60 (26–94) | 59 (26–94) | 62 (41–72) |
| No. age < 40 | 1 | 1 | 0 |
| Gender (n.s.) | |||
| Female | 10 (11) | 6 (9) | 4 (15) |
| Male | 84 (89) | 58 (91) | 22 (85) |
| Smoking (*) | |||
| No | 17 (18) | 7 (11) | 9 (35) |
| Yes | 76 (81) | 56 (88) | 17 (65) |
| NA | 1 (1) | 1 (2) | 0 |
| Alcohol usage (n.s.) | |||
| No | 38 (40) | 22 (34) | 14 (54) |
| yes | 55 (59) | 41 (64) | 12 (46) |
| NA | 1 (1) | 1 (2) | 0 |
| Tumour site (***) | |||
| base of the tongue | 16 (17) | 9 (14) | 7 (27) |
| hypopharynx | 4 (4) | 4 (6) | 0 |
| larynx | 21 (22) | 20 (31) | 0 |
| oral cavity | 21 (22) | 18 (28) | 1 (4) |
| oropharynx part | 9 (10) | 6 (9) | 2 (8) |
| tonsils | 23 (24) | 7 (11) | 16 (62) |
| Stage (n.s.) | |||
| I | 8 (9) | 6 (9) | 2 (8) |
| II | 8 (9) | 5 (8) | 3 (12) |
| III | 21 (22) | 17 (27) | 4 (15) |
| IV | 56 (60) | 35 (55) | 17 (65) |
| NA | 1 (1) | 1 (2) | 0 |
| Grade (*) | |||
| G1 | 19 (20) | 18 (28) | 1 (4) |
| G2 | 46 (49) | 26 (41) | 18 (69) |
| G3 | 28 (30) | 19 (30) | 7 (27) |
| G4 | 1 (1) | 1 (2) | 0 |
| Months of follow-up (median) (range) (n.s.) | 47 (0–139) | 29 (0–138) | 110 (16–139) |
| Five-Year Survival (%) | Univariate HR (95% CI), P | |||
|---|---|---|---|---|
| HPV(−) | HPV(+) | HPV(−) | HPV(+) | |
| CCND1 | ||||
| not | 38 | 82 | 1 | 1 |
| up | 29 | 0 | 1.04 (0.5–2.17), 0.92 | 46.95 (2.26–973.39), 0.01 |
| down | 0 | 77 | 2.6 (0.86–7.85), 0.09 | 2.88 (0.58–14.3), 0.2 |
| CCND2 | ||||
| not | 30 | 81 | 1 | 1 |
| up | 28 | 50 | 1.16 (0.59–2.29), 0.67 | 2.04 (0.54–7.69), 0.29 |
| down | 60 | 100 | 0.51 (0.15–1.72), 0.28 | NA |
| CCND3 | ||||
| not | 32 | 77 | 1 | 1 |
| up | 100 | 67 | NA | 0.9 (0.11–7.22), 0.92 |
| down | 0 | 0 | NA | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novotný, J.; Bandúrová, V.; Strnad, H.; Chovanec, M.; Hradilová, M.; Šáchová, J.; Šteffl, M.; Grušanović, J.; Kodet, R.; Pačes, V.; et al. Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas and Paired Normal Mucosae Reveals Cyclin D1 Deregulation and Compensatory Effect of Cyclin D2. Cancers 2020, 12, 792. https://doi.org/10.3390/cancers12040792
Novotný J, Bandúrová V, Strnad H, Chovanec M, Hradilová M, Šáchová J, Šteffl M, Grušanović J, Kodet R, Pačes V, et al. Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas and Paired Normal Mucosae Reveals Cyclin D1 Deregulation and Compensatory Effect of Cyclin D2. Cancers. 2020; 12(4):792. https://doi.org/10.3390/cancers12040792
Chicago/Turabian StyleNovotný, Jiří, Veronika Bandúrová, Hynek Strnad, Martin Chovanec, Miluše Hradilová, Jana Šáchová, Martin Šteffl, Josipa Grušanović, Roman Kodet, Václav Pačes, and et al. 2020. "Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas and Paired Normal Mucosae Reveals Cyclin D1 Deregulation and Compensatory Effect of Cyclin D2" Cancers 12, no. 4: 792. https://doi.org/10.3390/cancers12040792
APA StyleNovotný, J., Bandúrová, V., Strnad, H., Chovanec, M., Hradilová, M., Šáchová, J., Šteffl, M., Grušanović, J., Kodet, R., Pačes, V., Lacina, L., Smetana, K., Jr., Plzák, J., Kolář, M., & Vomastek, T. (2020). Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas and Paired Normal Mucosae Reveals Cyclin D1 Deregulation and Compensatory Effect of Cyclin D2. Cancers, 12(4), 792. https://doi.org/10.3390/cancers12040792