Aurora Borealis (Bora), Which Promotes Plk1 Activation by Aurora A, Has an Oncogenic Role in Ovarian Cancer

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
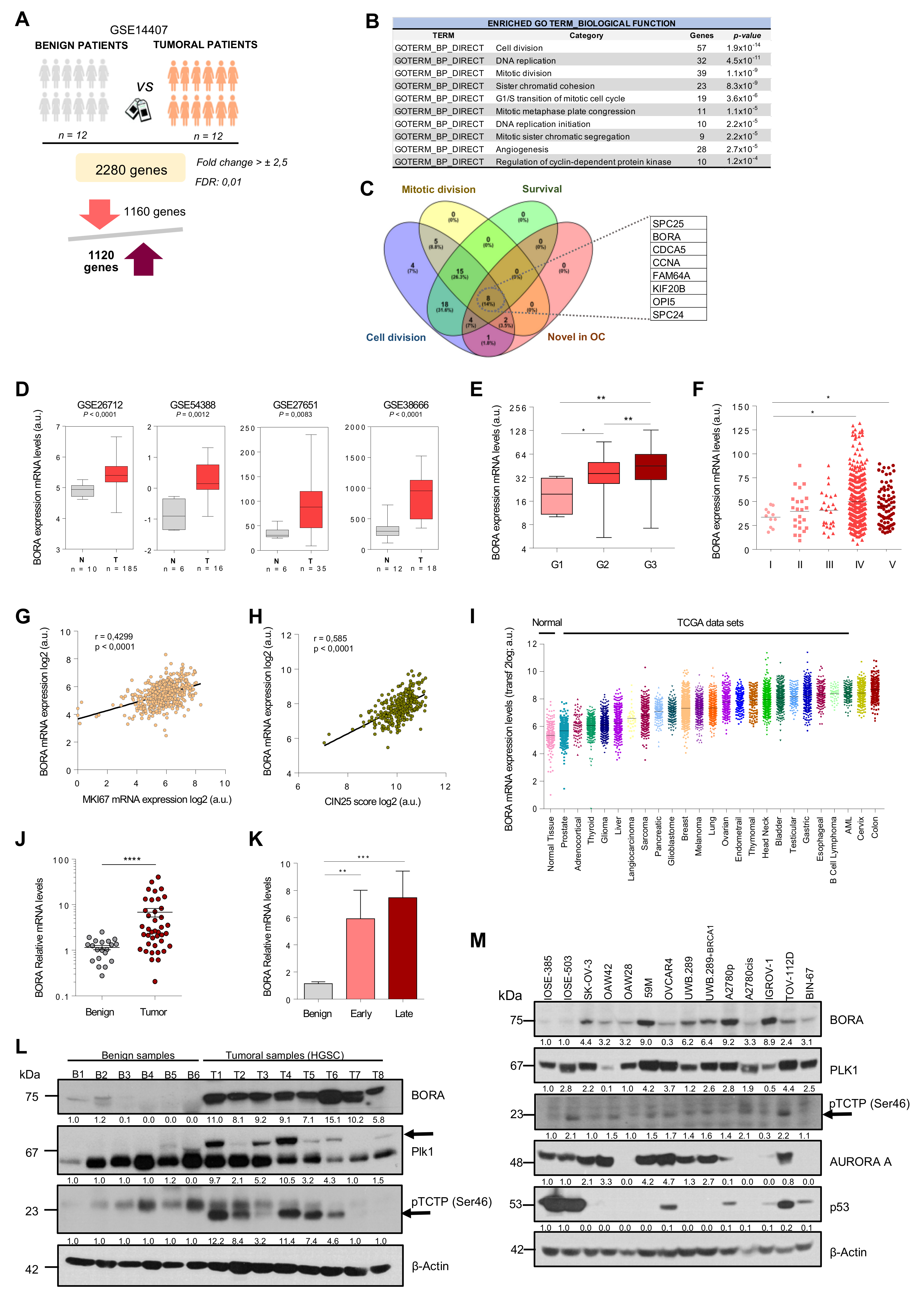
2.1. An Integrated Bioinformatics Screening Identifies Mitotic Regulators Potentially Involved in OC
2.2. BORA Overexpression Is Associated with Poor Prognosis in OC Patients
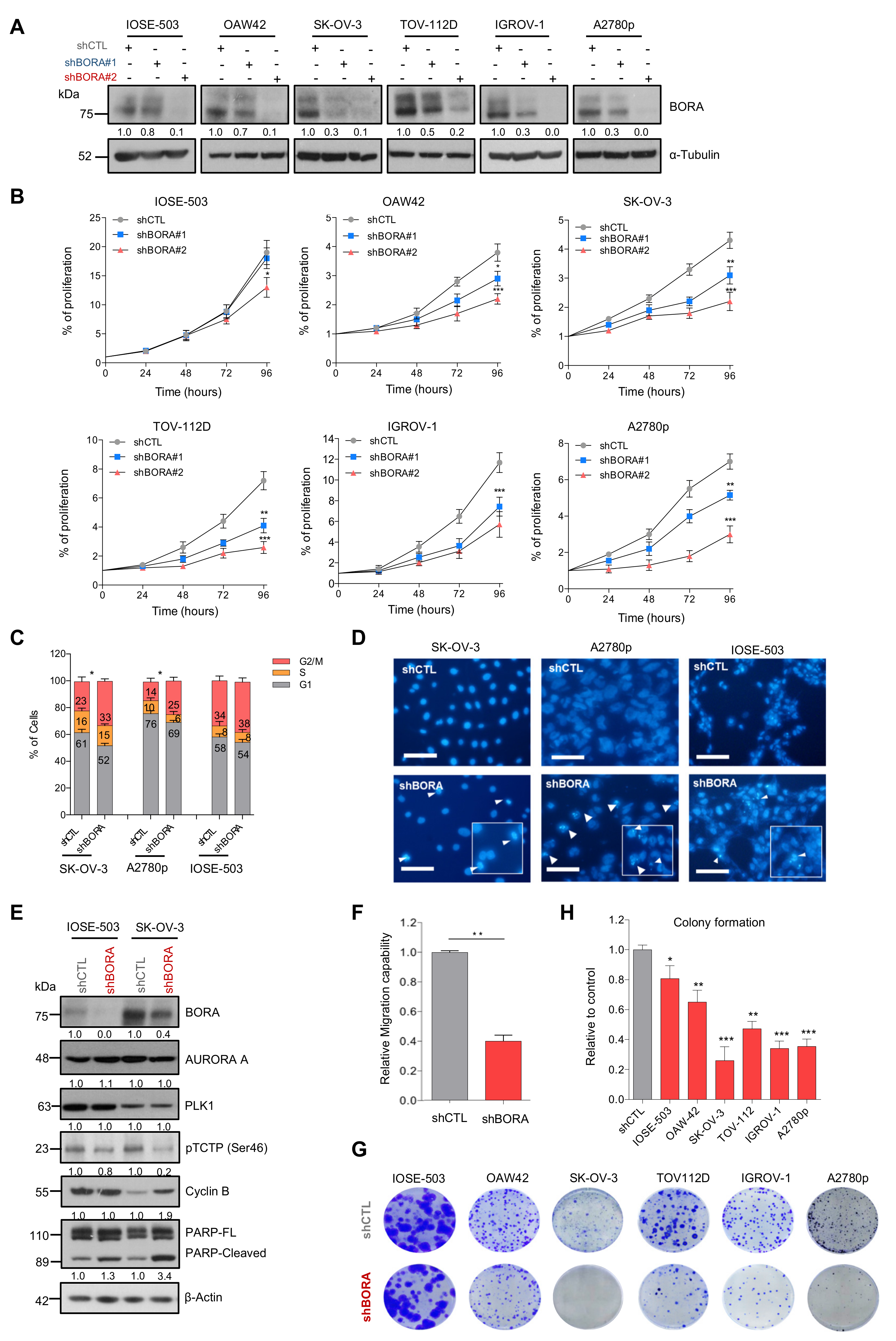
2.3. BORA Overexpression Renders Malignant Transformation Of Nontumoral Cells In Vitro
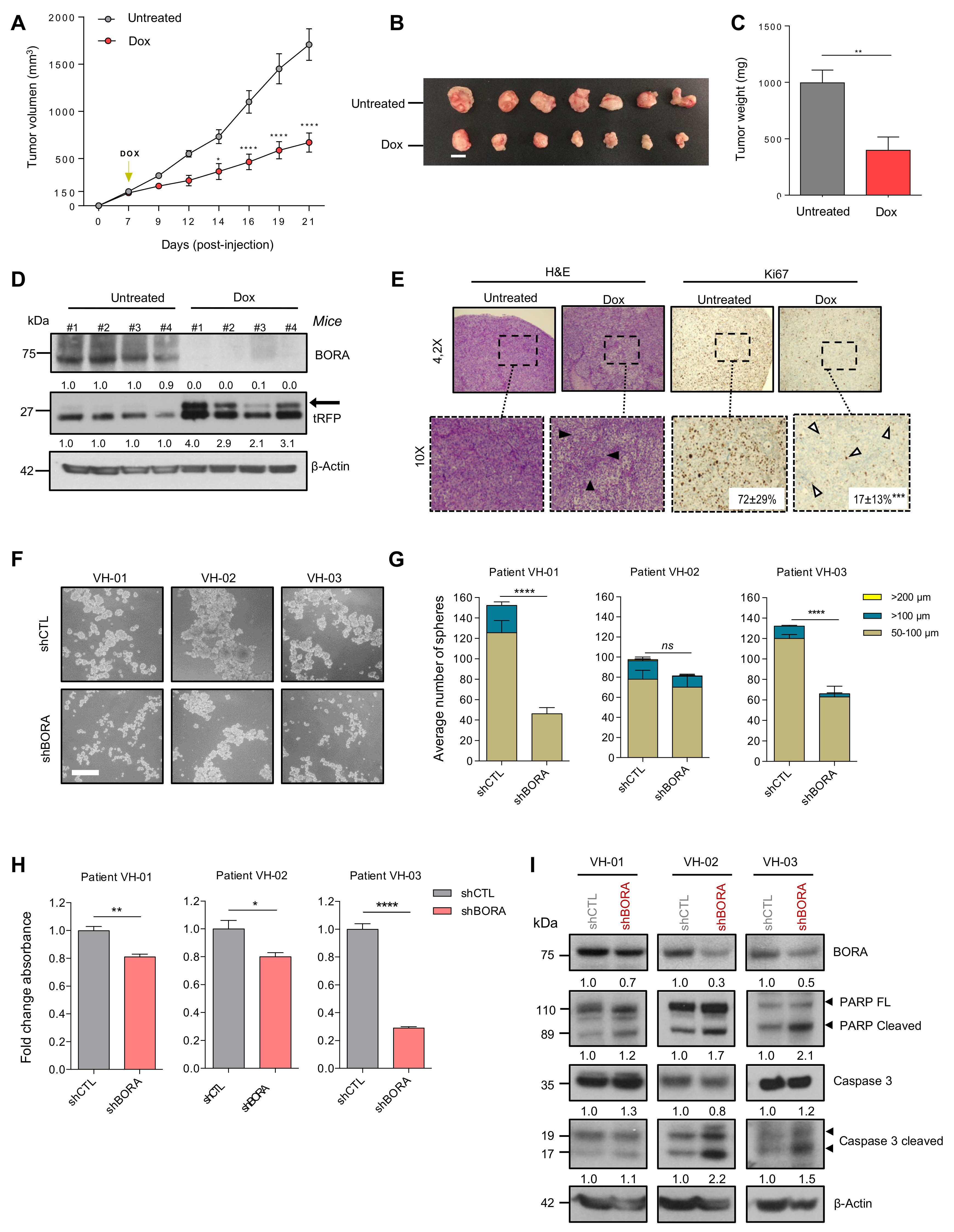
2.4. BORA Contributes to Ovarian Tumorigenesis In Vivo
2.5. BORA Silencing Has Tumor-Suppressive Effects In Vitro
2.6. BORA Depletion Impairs Tumor Growth In Vivo
2.7. BORA Silencing Reduces the Number and Viability of Patient-Derived Ascites Cells
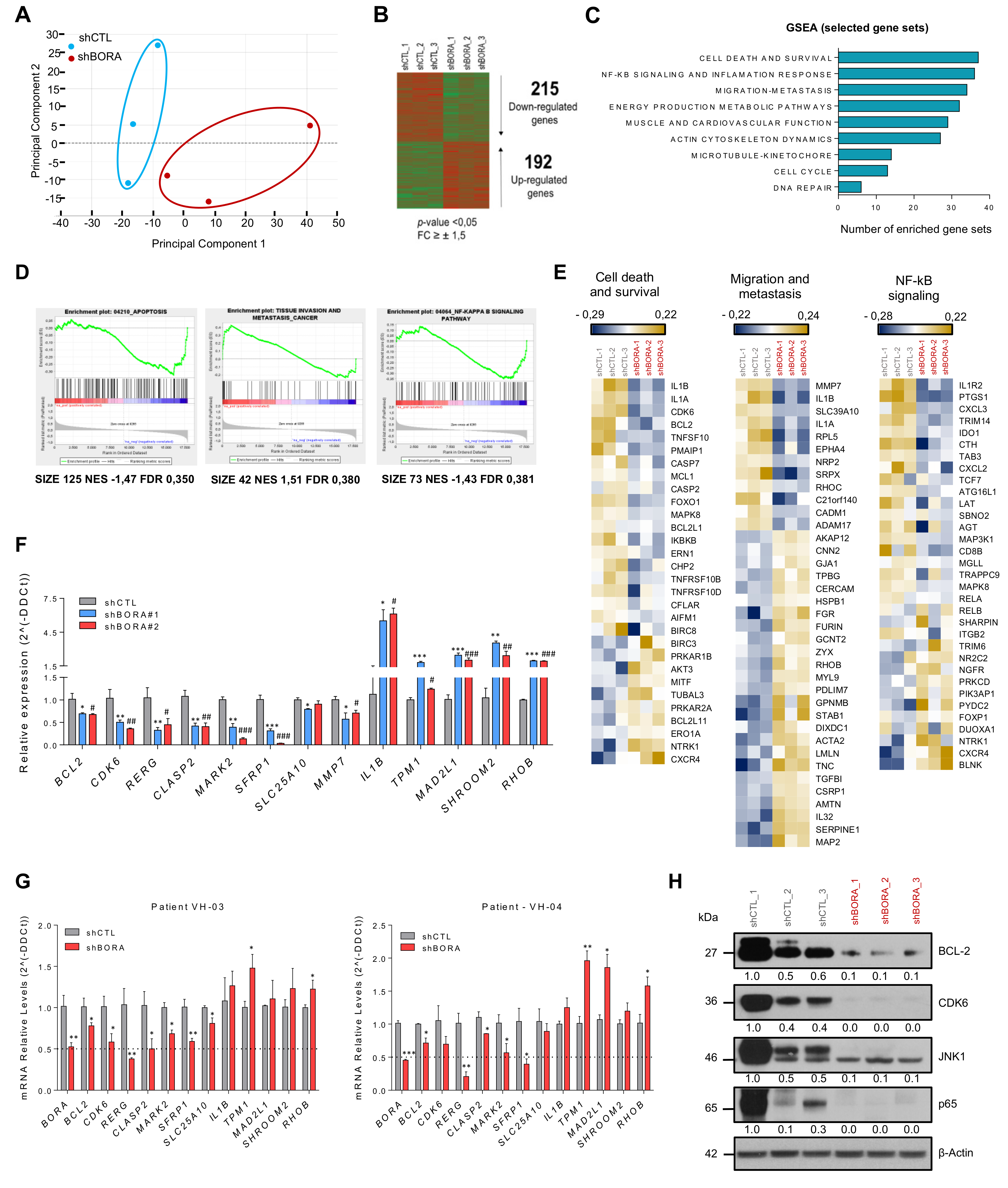
2.8. Reduction in BORA Levels Impact in Multiple Cancer-Related Genes
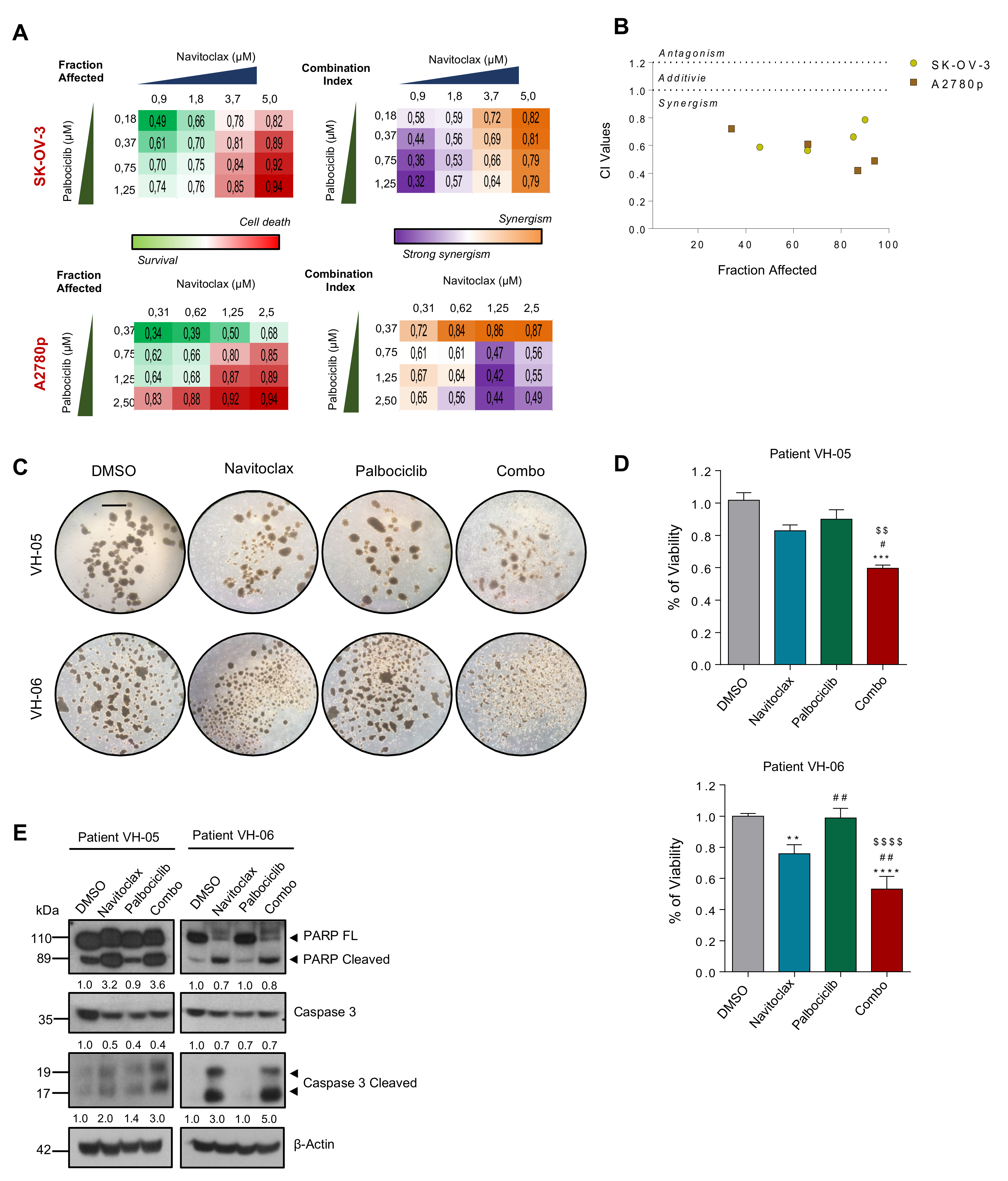
2.9. CDK6 and BCL2 Inhibitors Exert Synergistic Effect on OC Viability
3. Discussion
4. Material and Methods
4.1. Analysis of Cancer Genomic And Transcriptomic Datasets
4.2. Human Samples
4.3. Cell Lines
4.4. Protein Extraction, Immunoblot And Densiometry
4.5. Real-Time Quantitative PCR
4.6. Plasmids,Lentiviral Production and Generation of Stable Lines
4.7. Proliferation and Clonal Cell Growth Assays
4.8. Anchorage Independent Growth Assay (Soft Agar)
4.9. Boyden Chamber Migration Assay
4.10. Growth Inhibition Activity Assay
4.11. Fluorescence-Activated Cell Sorter Analysis (FACs)
4.12. Cell Death Assay
4.13. Mouse Xenograft
4.14. 3D Culture from Patient-Derived Ascitic Cells
4.15. Microarray Gene Expression Analysis
4.16. Drug Combination Studies
4.17. Immunohistochemistry
4.18. Statistical Methodologies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hutterer, A.; Berdnik, D.; Wirtz-Peitz, F.; Žigman, M.; Schleiffer, A.; Knoblich, J.A. Mitotic Activation of the Kinase Aurora-A Requires Its Binding Partner Bora. Dev. Cell 2006, 11, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.H.Y.; Santamaria, A.; Silljé, H.H.W.; Nigg, E.A. Plk1 regulates mitotic Aurora A function through βTrCP-dependent degradation of hBora. Chromosoma 2008, 117, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Gao, B.; Yu, J.; Yuan, J.; Lou, Z. ATR Regulates DNA Damage-Induced G2/M Checkpoint through the Aurora A Cofactor Bora. J. Biol. Chem. 2013, 288, 16139–16144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, Y.; Cirillo, L.; Panbianco, C.; Martino, L.; Tavernier, N.; Schwager, F.; van Hove, L.; Joly, N.; Santamaria, A.; Pintard, L.; et al. Cdk1 Phosphorylates SPAT-1/Bora to Promote Plk1 Activation in C. elegans and Human Cells. Cell Rep. 2016, 15, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrilla, A.; Cirillo, L.; Thomas, Y.; Gotta, M.; Pintard, L.; Santamaria, A. Mitotic entry: The interplay between Cdk1, Plk1 and Bora. Cell Cycle 2016, 4101, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, S.; Sundermann, L.; Labbé, J.C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 2018, 45, 637–650. [Google Scholar] [CrossRef]
- Zhang, Q.-X.; Gao, R.; Xiang, J.; Yuan, Z.-Y.; Qian, Y.-M.; Yan, M.; Wang, Z.-F.; Liu, Q.; Zhao, H.-D.; Liu, C.-H. Cell cycle protein bora serves as a novel poor prognostic factor in multiple adenocarcinomas. Oncotarget 2017, 8, 43838–43852. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Vasey, P.A.; Jayson, G.C.; Gordon, A.; Gabra, H.; Coleman, R.; Atkinson, R.; Parkin, D.; Paul, J.; Hay, A.; Kaye, S.B. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel-carboplatin as first-line chemotherpy for ovarian carcinoma. J. Natl. Cancer Inst. 2004, 96, 1682–1691. [Google Scholar] [CrossRef] [Green Version]
- Harter, P.; Sehouli, J.; Reuss, A.; Hasenburg, A.; Scambia, G.; Cibula, D.; Mahner, S.; Vergote, I.; Reinthaller, A.; Burges, A.; et al. Prospective validation study of a predictive score for operability of recurrent ovarian cancer: The multicenter intergroup study DESKTOP II. A project of the AGO kommission OVAR, AGO study group, NOGGO, AGO-Austria, and MITO. Int. J. Gynecol. Cancer 2011, 21, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cáncer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- George, A.; Kaye, S.; Banerjee, S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cáncer. Nat. Rev. Clin. Oncol. 2017, 14, 284–296. [Google Scholar] [CrossRef] [PubMed]
- de Jaeghere, E.; Vandecasteele, K.; Claes, K.; Makar, A.; Tummers, P.; Cocquyt, V.; Denys, H. Incorporating PARP-inhibitors into clinical routine: A tailored treatment strategy to tackle ovarian cáncer. Acta Clin. Belgica Int. J. Clin. Lab. Med. 2017, 72, 6–11. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.L.; Jeusset, L.M.P.; Lepage, C.C.; McManus, K.J. Evolving therapeutic strategies to exploit chromosome instability in cáncer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Di Rora, A.G.; Iacobucci, I.; Martinelli, G. The cell cycle checkpoint inhibitors in the treatment of leukemias. J. Hematol. Oncol. 2017, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM–CHK2 and ATR–CHK1 pathways for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöffski, P.; Awada, A.; Dumez, H.; Gil, T.; Bartholomeus, S.; Wolter, P.; Taton, M.; Fritsch, H.; Glomb, P.; Munzert, G. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 2012, 48, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patientswith tp53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bossche, J.; Lardon, F.; Deschoolmeester, V.; de Pauw, I.; Vermorken, J.B.; Specenier, P.; Pauwels, P.; Peeters, M.; Wouters, A. Spotlight on Volasertib: Preclinical and Clinical Evaluation of a Promising Plk1 Inhibitor. Med. Res. Rev. 2016, 36, 749–786. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Selle, F.; Weber, B.; Ray-Coquard, I.L.; Vergote, I.; Sufliarsky, J.; del Campo, J.M.; Lortholary, A.; Lesoin, A.; Follana, P.; et al. Volasertib versus chemotherapy in platinum-resistant or-refractory ovarian cancer: A randomized phase II groupe des investigateurs nationaux pour l’etude des cancers de l’ovaire study. J. Clin. Oncol. 2016, 34, 706–713. [Google Scholar] [CrossRef]
- Döhner, H.; Lübbert, M.; Fiedler, W.; Fouillard, L.; Haaland, A.; Brandwein, J.M.; Lepretre, S.; Reman, O.; Turlure, P.; Ottmann, O.G.; et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood 2014, 124, 1426–1433. [Google Scholar] [CrossRef]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Rath, O.; Kozielski, F. Kinesins and cáncer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef]
- Niu, N.; Qin, Y.; Fridley, B.L.; Hou, J.; Kalari, K.R.; Zhu, M.; Wu, T.Y.; Jenkins, G.D.; Batzler, A.; Wang, L. Radiation pharmacogenomics: A genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines. Genome Res. 2010, 20, 1482–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Cucchi, U.; Gianellini, L.M.; de Ponti, A.; Sola, F.; Alzani, R.; Patton, V.; Pezzoni, A.; Troiani, S.; Saccardo, M.B.; Rizzi, S.; et al. Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res. 2010, 30, 4973–4986. [Google Scholar] [PubMed]
- Cifone, M.A.; Fidler, I.J. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc. Natl. Acad. Sci. USA 1980, 77, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, R.; Narisawa-Saito, M.; Yugawa, T.; Fujita, M.; Tashiro, H.; Katabuchi, H.; Kiyono, T. Oncogenic transformation of human ovarian surface epithelial cells with defined cellular oncogenes. Carcinogenesis 2009, 30, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Bowles, D.W.; Diamond, J.R.; Lam, E.T.; Weekes, C.D.; Astling, D.P.; Anderson, R.T.; Leong, S.; Gore, L.; Varella-Garcia, M.; Vogler, B.W.; et al. Phase I study of oral rigosertib (ON 01910.NA), a dual inhibitor of the PI3K and PLK1 pathways, in adult patients with advanced solid malignancies. Clin. Cancer Res. 2014, 20, 1656–1665. [Google Scholar] [CrossRef] [Green Version]
- Seki, A.; Coppinger, J.A.; Du, H.; Jang, C.Y.; Yates, J.R.; Fang, G. Plk1- and β-TrCP-dependent degradation of Bora controls mitotic progression. J. Cell Biol. 2008, 181, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Bruinsma, W.; Macůrek, L.; Freire, R.; Lindqvist, A.; Medema, R.H. Bora and Aurora-A continue to activate Plk1 in mitosis. J. Cell Sci. 2014, 127, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.P.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Szender, J.B.; Emmons, T.; Belliotti, S.; Dickson, D.; Khan, A.; Morrell, K.; Khan, A.N.M.N.; Singel, K.L.; Mayor, P.C.; Moysich, K.B.; et al. Impact of ascites volume on clinical outcomes in ovarian cancer: A cohort study. Gynecol. Oncol. 2017, 146, 491–497. [Google Scholar] [CrossRef]
- Kipps, E.; Tan, D.S.P.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, T.G.; Thériault, B.L.; Campbell, E.J.; Nachtigal, M.W. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat. Protoc. 2007, 1, 2643–2649. [Google Scholar] [CrossRef] [PubMed]
- Majem, B.; Parrilla, A.; Jiménez, C.; Suárez-Cabrera, L.; Barber, M.; Marín, A.; Castellví, J.; Tamayo, G.; Moreno-Bueno, G.; Ponce, J.; et al. MicroRNA-654-5p suppresses ovarian cancer development impacting on MYC, WNT and AKT pathways. Oncogene 2019, 38, 6035–6050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA approval: Palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cáncer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Wilson, M.L.; Hamanaka, R.; Chase, D.; Kung, H.F.; Longo, D.L.; Ferris, D.K. Malignant transformation of mammalian cells initiated by constitutive expression of the Polo-like kinase. Biochem. Biophys. Res. Commun. 1997, 234, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ivanov, A.I.; Fisher, P.B.; Fu, Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. eLife 2016, 5, e10734. [Google Scholar] [CrossRef]
- Wang, J.; Hu, K.; Guo, J.; Cheng, F.; Lv, J.; Jiang, W.; Lu, W.; Liu, J.; Pang, X.; Liu, M. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat. Commun. 2016, 7, 11363. [Google Scholar] [CrossRef] [Green Version]
- Noack, S.; Raab, M.; Matthess, Y.; Sanhaji, M.; Krämer, A.; Gyorffy, B.; Kaderali, L.; El-Balat, A.; Becker, S.; Strebhardt, K. Synthetic lethality in CCNE1-amplified high grade serous ovarian cancer through combined inhibition of polo-like kinase 1 and microtubule dynamics. Oncotarget 2018, 9, 25842–25859. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, G.; Gao, Z.; Li, S.; Li, Z.; Bi, J.; Liu, X.; Li, Z.; Kong, C. Comprehensive analysis of differentially expressed genes associated with PLK1 in bladder cáncer. BMC Cancer 2017, 17, 861. [Google Scholar] [CrossRef] [Green Version]
- Takaki, T.; Trenz, K.; Costanzo, V.; Petronczki, M. Polo-like kinase 1 reaches beyond mitosis-cytokinesis, DNA damage response, and development. Curr. Opin. Cell Biol. 2008, 20, 650–660. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, G.; Wachowicz, P.; Martínez-Martínez, S.; Oller, J.; Méndez-Barbero, N.; Escobar, B.; González-Loyola, A.; Takaki, T.; el Bakkali, A.; Cámara, J.A.; et al. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat. Med. 2017, 23, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Roberts, A.W.; Colman, P.M.; Adams, J.M. Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer 2016, 2, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Su, J.; Jiao, B.; Shen, L.; Ma, L.; Qu, X.; Yu, C.; Jiang, X.; Xu, Y.; Sun, L. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int. J. Oncol. 2016, 49, 2507–2519. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, A.; Sonego, M.; Pellizzari, I.; Pellarin, I.; Canzonieri, V.; D’Andrea, S.; Benevol, S.; Sorio, R.; Giorda, G.; Califano, D.; et al. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol. Med. 2017, 9, 1415–1433. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- de Dominici, M.; Porazzi, P.; Soliera, A.R.; Mariani, S.A.; Addya, S.; Fortina, P.; Peterson, L.F.; Spinelli, O.; Rambaldi, A.; Martinelli, G.; et al. Targeting CDK6 and BCL2 exploits the “MYB addiction” of Ph þ acute lymphoblastic leukemia. Cancer Res. 2018, 78, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jubierre, L.; Soriano, A.; Planells-Ferrer, L.; París-Coderch, L.; Tenbaum, S.P.; Romero, O.A.; Moubarak, R.S.; Almazán-Moga, A.; Molist, C.; Roma, J.; et al. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene 2016, 35, 5179–5190. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrilla, A.; Barber, M.; Majem, B.; Castellví, J.; Morote, J.; Sánchez, J.L.; Pérez-Benavente, A.; Segura, M.F.; Gil-Moreno, A.; Santamaria, A. Aurora Borealis (Bora), Which Promotes Plk1 Activation by Aurora A, Has an Oncogenic Role in Ovarian Cancer. Cancers 2020, 12, 886. https://doi.org/10.3390/cancers12040886
Parrilla A, Barber M, Majem B, Castellví J, Morote J, Sánchez JL, Pérez-Benavente A, Segura MF, Gil-Moreno A, Santamaria A. Aurora Borealis (Bora), Which Promotes Plk1 Activation by Aurora A, Has an Oncogenic Role in Ovarian Cancer. Cancers. 2020; 12(4):886. https://doi.org/10.3390/cancers12040886
Chicago/Turabian StyleParrilla, Alfonso, Marta Barber, Blanca Majem, Josep Castellví, Juan Morote, José Luis Sánchez, Asunción Pérez-Benavente, Miguel F. Segura, Antonio Gil-Moreno, and Anna Santamaria. 2020. "Aurora Borealis (Bora), Which Promotes Plk1 Activation by Aurora A, Has an Oncogenic Role in Ovarian Cancer" Cancers 12, no. 4: 886. https://doi.org/10.3390/cancers12040886
APA StyleParrilla, A., Barber, M., Majem, B., Castellví, J., Morote, J., Sánchez, J. L., Pérez-Benavente, A., Segura, M. F., Gil-Moreno, A., & Santamaria, A. (2020). Aurora Borealis (Bora), Which Promotes Plk1 Activation by Aurora A, Has an Oncogenic Role in Ovarian Cancer. Cancers, 12(4), 886. https://doi.org/10.3390/cancers12040886