Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes

 , ,
, ,
Abstract
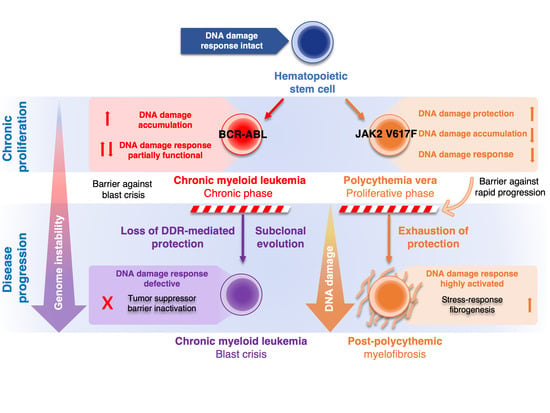
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of DDR in CML and PV
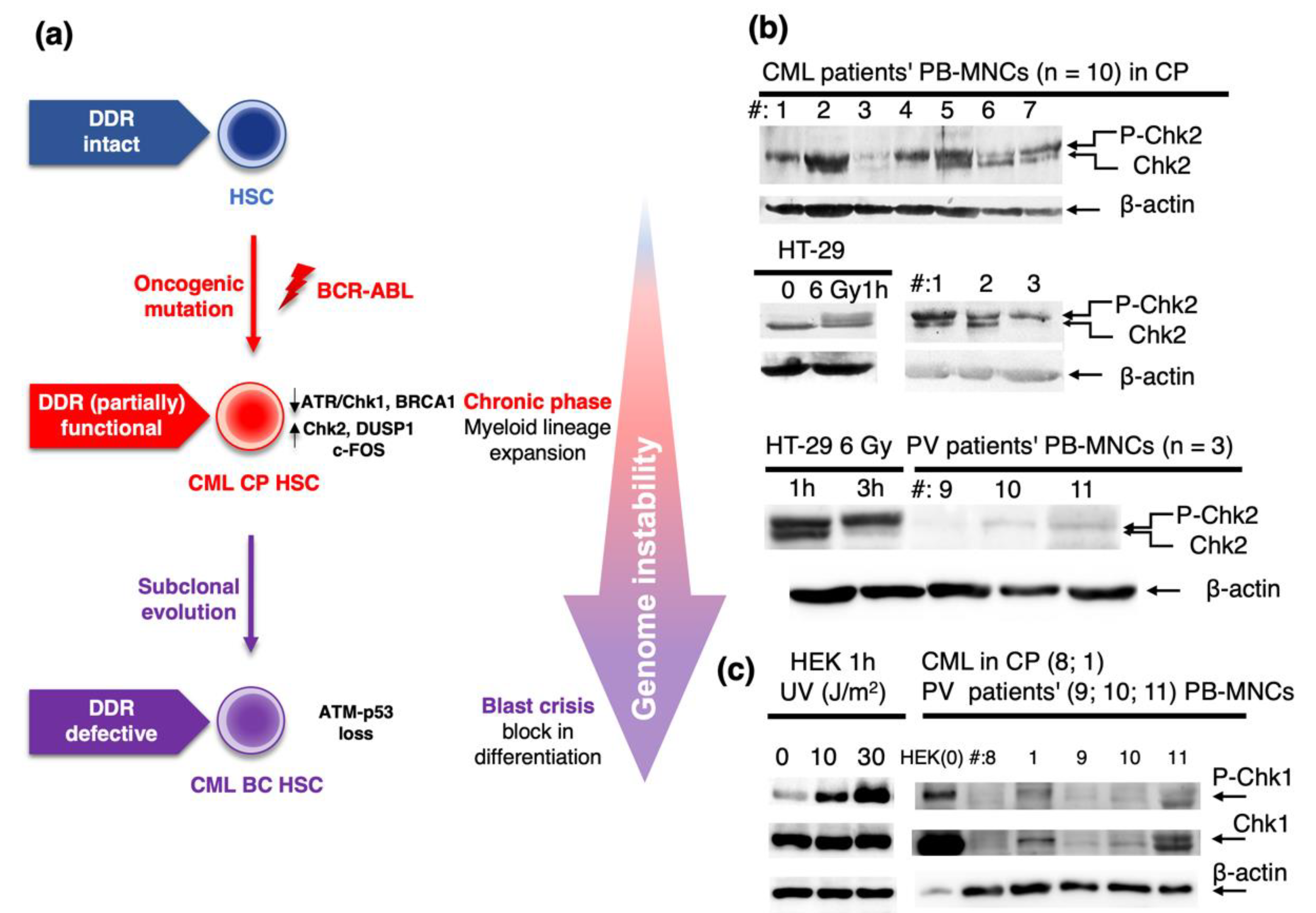
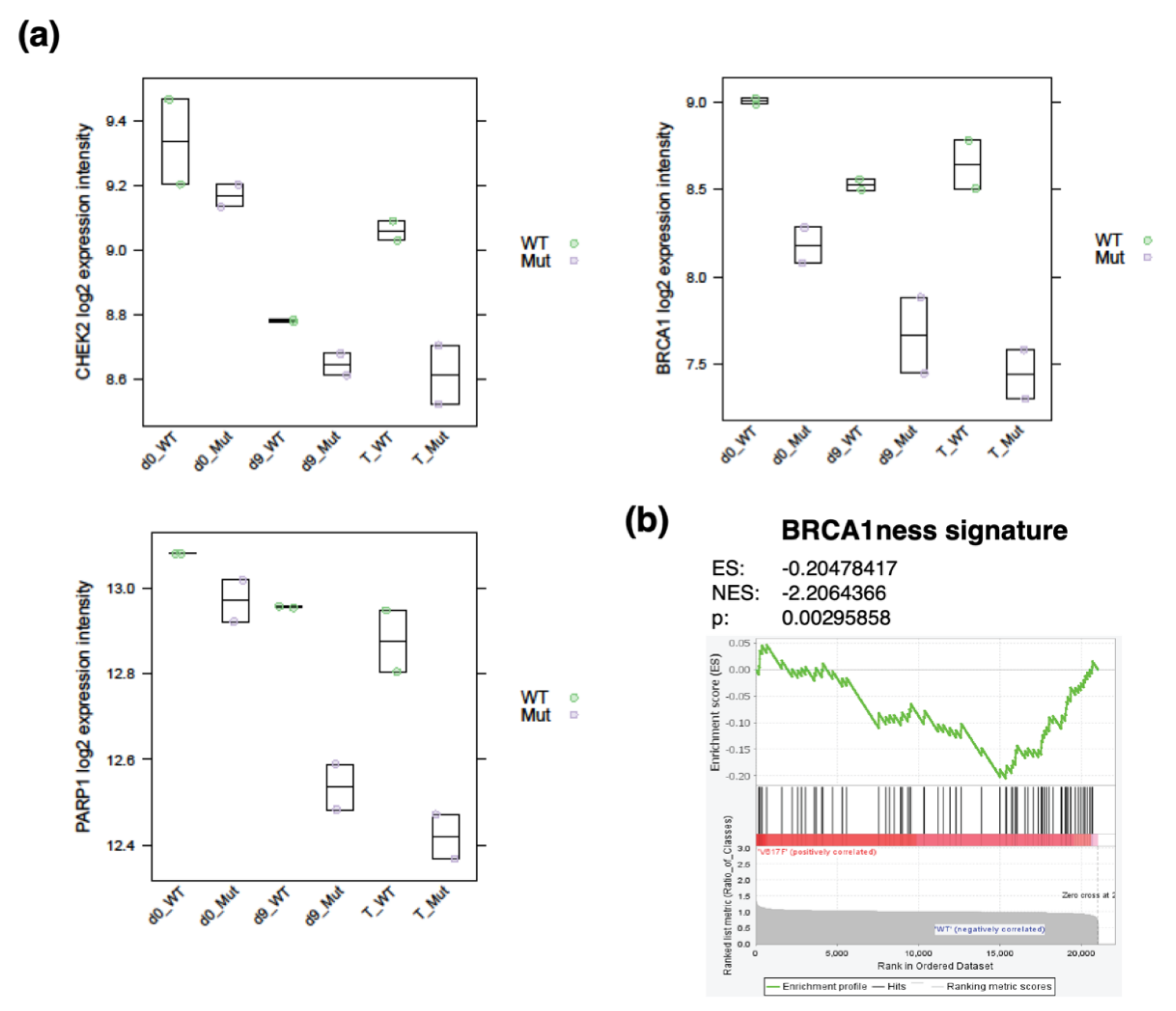
2.1. Role of DDR in Chronic Phase of CML and Progression to Blast Crisis
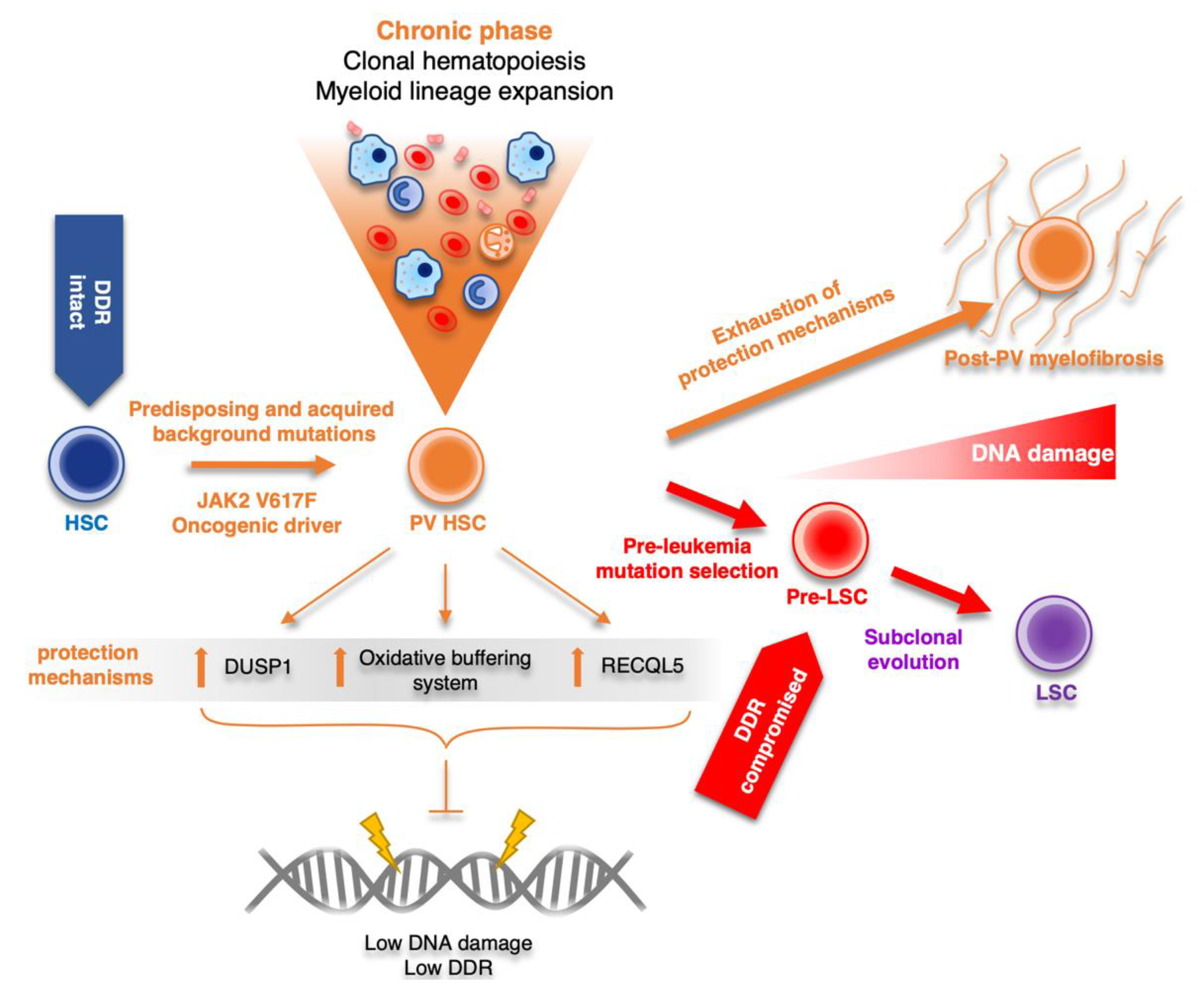
2.2. Role of DDR in Polycythemia Vera
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Methodology for Obtaining Previously Unpublished Data
Appendix A.2. Patients
References
- de Klein, A.; van Kessel, A.G.; Grosveld, G.; Bartram, C.R.; Hagemeijer, A.; Bootsma, D.; Spurr, N.K.; Heisterkamp, N.; Groffen, J.; Stephenson, J.R. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1982, 300, 765–767. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Mirantes, C.; Passegué, E.; Pietras, E.M. Pro-inflammatory cytokines: Emerging players regulating HSC function in normal and diseased hematopoiesis. Exp. Cell Res. 2014, 329, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediat. Inflamm. 2015, 2015, e102476. [Google Scholar] [CrossRef] [Green Version]
- Craver, B.M.; El Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers (Basel) 2018, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Reynaud, D.; Pietras, E.; Barry-Holson, K.; Mir, A.; Binnewies, M.; Jeanne, M.; Sala-Torra, O.; Radich, J.P.; Passegué, E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011, 20, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.-U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welner, R.S.; Amabile, G.; Bararia, D.; Czibere, A.; Yang, H.; Zhang, H.; Pontes, L.L.D.F.; Ye, M.; Levantini, E.; Di Ruscio, A.; et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell 2015, 27, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2018, 33, 29–43.e7. [Google Scholar] [CrossRef] [Green Version]
- Mendez Luque, L.F.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression. and Symptoms. Curr. Hematol. Malig. Rep. 2019, 14, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.-T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [Green Version]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar] [CrossRef]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takacova, S.; Slany, R.; Bartkova, J.; Stranecky, V.; Dolezel, P.; Luzna, P.; Bartek, J.; Divoky, V. DNA damage response and inflammatory signaling limit the MLL-ENL-induced leukemogenesis in vivo. Cancer Cell 2012, 21, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, M.T.; So, C.W.E. DNA damage accumulation and repair defects in acute myeloid leukemia: Implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014, 123, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Nilles, N.; Fahrenkrog, B. Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Gilliland, D.G. Oncogenes in myeloproliferative disorders. Cell Cycle 2007, 6, 550–566. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Wasik, M.A.; Slupianek, A.; Salomoni, P.; Kitamura, T.; Calabretta, B.; Skorski, T. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med. 1999, 189, 1229–1242. [Google Scholar] [CrossRef]
- Walz, C.; Cross, N.C.P.; Van Etten, R.A.; Reiter, A. Comparison of mutated ABL1 and JAK2 as oncogenes and drug targets in myeloproliferative disorders. Leukemia 2008, 22, 1320–1334. [Google Scholar] [CrossRef] [Green Version]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; Van Etten, R.A. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef]
- Konopka, J.B.; Watanabe, S.M.; Singer, J.W.; Collins, S.J.; Witte, O.N. Cell lines and clinical isolates derived from Ph1-positive chronic myelogenous leukemia patients express c-abl proteins with a common structural alteration. Proc. Natl. Acad. Sci. USA 1985, 82, 1810–1814. [Google Scholar] [CrossRef] [Green Version]
- Konopka, J.B.; Witte, O.N. Activation of the abl oncogene in murine and human leukemias. Biochim. Biophys. Acta 1985, 823, 1–17. [Google Scholar] [CrossRef]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef]
- Gregor, T.; Bosakova, M.K.; Nita, A.; Abraham, S.P.; Fafilek, B.; Cernohorsky, N.H.; Rynes, J.; Foldynova-Trantirkova, S.; Zackova, D.; Mayer, J.; et al. Elucidation of protein interactions necessary for the maintenance of the BCR-ABL signaling complex. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, C.; Li, D.; Li, S. Molecular and cellular bases of chronic myeloid leukemia. Protein Cell 2010, 1, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmes, S.; Hafer, M.; Vuorio, J.; Tucker, J.A.; Winkelmann, H.; Löchte, S.; Stanly, T.A.; Pulgar Prieto, K.D.; Poojari, C.; Sharma, V.; et al. Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science 2020, 367, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Green, A.R. The myeloproliferative disorders. N. Engl. J. Med. 2006, 355, 2452–2466. [Google Scholar] [CrossRef]
- Calabretta, B.; Perrotti, D. The biology of CML blast crisis. Blood 2004, 103, 4010–4022. [Google Scholar] [CrossRef] [Green Version]
- Popp, H.D.; Kohl, V.; Naumann, N.; Flach, J.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Saussele, S.; Hofmann, W.-K.; et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 1177. [Google Scholar] [CrossRef] [Green Version]
- Dierov, J.; Dierova, R.; Carroll, M. BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell 2004, 5, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Nieborowska-Skorska, M.; Stoklosa, T.; Datta, M.; Czechowska, A.; Rink, L.; Slupianek, A.; Koptyra, M.; Seferynska, I.; Krszyna, K.; Blasiak, J.; et al. ATR-Chk1 axis protects BCR/ABL leukemia cells from the lethal effect of DNA double-strand breaks. Cell Cycle 2006, 5, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Shafman, T.; Khanna, K.K.; Kedar, P.; Spring, K.; Kozlov, S.; Yen, T.; Hobson, K.; Gatei, M.; Zhang, N.; Watters, D.; et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature 1997, 387, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Wood, L.D.; Whitaker, L.L.; Canman, C.E.; Morgan, S.E.; Xu, Y.; Barlow, C.; Baltimore, D.; Wynshaw-Boris, A.; Kastan, M.B.; et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997, 387, 516–519. [Google Scholar] [CrossRef]
- Wetzler, M.; Talpaz, M.; Van Etten, R.A.; Hirsh-Ginsberg, C.; Beran, M.; Kurzrock, R. Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation. J. Clin. Investig. 1993, 92, 1925–1939. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Sato, M.; Piao, J.; Miyamoto, S.; Isoda, T.; Kitagawa, M.; Honda, H.; Mizutani, S. ATM-dependent DNA damage-response pathway as a determinant in chronic myelogenous leukemia. DNA Repair (Amst.) 2013, 12, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Xu, X.; Tsvetkov, L.M.; Stern, D.F. Chk2 activation and phosphorylation-dependent oligomerization. Mol. Cell. Biol. 2002, 22, 4419–4432. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.; Jarrousse, S.; Buet, D.; Dugray, A.; Bonnet, M.-L.; Vozenin-Brotons, M.-C.; Guilhot, F.; Turhan, A.G.; Feunteun, J.; Bourhis, J. Down-regulation of BRCA1 in BCR-ABL-expressing hematopoietic cells. Blood 2003, 101, 4583–4588. [Google Scholar] [CrossRef] [Green Version]
- Dkhissi, F.; Aggoune, D.; Pontis, J.; Sorel, N.; Piccirilli, N.; LeCorf, A.; Guilhot, F.; Chomel, J.-C.; Ait-Si-Ali, S.; Turhan, A.G. The downregulation of BAP1 expression by BCR-ABL reduces the stability of BRCA1 in chronic myeloid leukemia. Exp. Hematol. 2015, 43, 775–780. [Google Scholar] [CrossRef]
- Huen, M.S.Y.; Sy, S.M.H.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slupianek, A.; Poplawski, T.; Jozwiakowski, S.K.; Cramer, K.; Pytel, D.; Stoczynska, E.; Nowicki, M.O.; Blasiak, J.; Skorski, T. BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res. 2011, 71, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canitrot, Y.; Laurent, G.; Astarie-Dequeker, C.; Bordier, C.; Cazaux, C.; Hoffmann, J.-S. Enhanced expression and activity of DNA polymerase beta in chronic myelogenous leukemia. Anticancer Res. 2006, 26, 523–525. [Google Scholar] [PubMed]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.P.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef] [Green Version]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar]
- Fernandes, M.S.; Reddy, M.M.; Gonneville, J.R.; DeRoo, S.C.; Podar, K.; Griffin, J.D.; Weinstock, D.M.; Sattler, M. BCR-ABL promotes the frequency of mutagenic single-strand annealing DNA repair. Blood 2009, 114, 1813–1819. [Google Scholar] [CrossRef] [Green Version]
- Dierov, J.; Sanchez, P.; Burke, B.; Padilla-Nash, H.; Putt, M.; Ried, T.; Carroll, M. BCR/ABL induces chromosomal instability after genotoxic stress and alters the cell death threshold. Leukemia 2009, 23, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Nieborowska-Skorska, M.; Sullivan, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Hoser, G.; Maifrede, S.; Martinez, E.; Di Marcantonio, D.; Bolton-Gillespie, E.; Cramer-Morales, K.; et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J. Clin. Investig. 2017, 127, 2392–2406. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef]
- Bourgeais, J.; Ishac, N.; Medrzycki, M.; Brachet-Botineau, M.; Desbourdes, L.; Gouilleux-Gruart, V.; Pecnard, E.; Rouleux-Bonnin, F.; Gyan, E.; Domenech, J.; et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget 2017, 8, 41876–41889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.; Lee, S.-R.; Yang, K.-S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.-C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Rudolph, J. Redox regulation of MAP kinase phosphatase 3. Biochemistry 2006, 45, 8476–8487. [Google Scholar] [CrossRef]
- Naughton, R.; Quiney, C.; Turner, S.D.; Cotter, T.G. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia 2009, 23, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Kesarwani, M.; Kincaid, Z.; Gomaa, A.; Huber, E.; Rohrabaugh, S.; Siddiqui, Z.; Bouso, M.F.; Latif, T.; Xu, M.; Komurov, K.; et al. c-Fos and Dusp1 confer non-oncogene addiction in BCR-ABL induced leukemia. Nat. Med. 2017, 23, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Liu, L.; Levin, D.E. Stressing out or stressing in: Intracellular pathways for SAPK activation. Curr. Genet. 2019, 65, 417–421. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in tumorigenesis, tumor progression and therapy. Cancer Med. 2016, 5, 2061–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.P.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xL Deamidation Pathway in Myeloproliferative Disorders. N. Engl. J. Med. 2008, 359, 2778–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef]
- Li, J.; Spensberger, D.; Ahn, J.S.; Anand, S.; Beer, P.A.; Ghevaert, C.; Chen, E.; Forrai, A.; Scott, L.M.; Ferreira, R.; et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood 2010, 116, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Plo, I.; Nakatake, M.; Malivert, L.; de Villartay, J.-P.; Giraudier, S.; Villeval, J.-L.; Wiesmuller, L.; Vainchenker, W. JAK2 stimulates homologous recombination and genetic instability: Potential implication in the heterogeneity of myeloproliferative disorders. Blood 2008, 112, 1402–1412. [Google Scholar] [CrossRef]
- Chen, E.; Ahn, J.S.; Massie, C.E.; Clynes, D.; Godfrey, A.L.; Li, J.; Park, H.J.; Nangalia, J.; Silber, Y.; Mullally, A.; et al. JAK2V617F promotes replication fork stalling with disease-restricted impairment of the intra-S checkpoint response. Proc. Natl. Acad. Sci. USA 2014, 111, 15190–15195. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Harutyunyan, A.; Berg, T.; Gisslinger, B.; Schalling, M.; Bagienski, K.; Olcaydu, D.; Passamonti, F.; Rumi, E.; Pietra, D.; et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood 2011, 118, 167–176. [Google Scholar] [CrossRef]
- Kojima, H.; Kunimoto, H.; Inoue, T.; Nakajima, K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle 2012, 11, 730–739. [Google Scholar] [CrossRef]
- Sattler, M.; Winkler, T.; Verma, S.; Byrne, C.H.; Shrikhande, G.; Salgia, R.; Griffin, J.D. Hematopoietic growth factors signal through the formation of reactive oxygen species. Blood 1999, 93, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Stetka, J.; Vyhlidalova, P.; Lanikova, L.; Koralkova, P.; Gursky, J.; Hlusi, A.; Flodr, P.; Hubackova, S.; Bartek, J.; Hodny, Z.; et al. Addiction to DUSP1 protects JAK2V617F-driven polycythemia vera progenitors against inflammatory stress and DNA damage, allowing chronic proliferation. Oncogene 2019, 38, 5627–5642. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Horibe, S.; Takagi, M.; Unno, J.; Nagasawa, M.; Morio, T.; Arai, A.; Miura, O.; Ohta, M.; Kitagawa, M.; Mizutani, S. DNA damage check points prevent leukemic transformation in myelodysplastic syndrome. Leukemia 2007, 21, 2195–2198. [Google Scholar] [CrossRef]
- Boehrer, S.; Adès, L.; Tajeddine, N.; Hofmann, W.K.; Kriener, S.; Bug, G.; Ottmann, O.G.; Ruthardt, M.; Galluzzi, L.; Fouassier, C.; et al. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene 2009, 28, 2205–2218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Walker, C.L. Differential localization of ATM is correlated with activation of distinct downstream signaling pathways. Cell Cycle 2010, 9, 3685–3686. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, S.V.; Waardenberg, A.J.; Engholm-Keller, K.; Arthur, J.W.; Graham, M.E.; Lavin, M. Reactive Oxygen Species (ROS)-Activated ATM-Dependent Phosphorylation of Cytoplasmic Substrates Identified by Large-Scale Phosphoproteomics Screen. Mol. Cell Proteomics 2016, 15, 1032–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Ahn, J.S.; Sykes, D.B.; Breyfogle, L.J.; Godfrey, A.L.; Nangalia, J.; Ko, A.; DeAngelo, D.J.; Green, A.R.; Mullally, A. RECQL5 Suppresses Oncogenic JAK2-Induced Replication Stress and Genomic Instability. Cell Rep. 2015, 13, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Severson, T.M.; Wolf, D.M.; Yau, C.; Peeters, J.; Wehkam, D.; Schouten, P.C.; Chin, S.-F.; Majewski, I.J.; Michaut, M.; Bosma, A.; et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. 2017, 19, 99. [Google Scholar] [CrossRef] [PubMed]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef] [Green Version]
- Iwasa, H.; Han, J.; Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003, 8, 131–144. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, W.; Li, Y.; Berenzon, D.; Wang, X.; Wang, J.; Mascarenhas, J.; Xu, M.; Hoffman, R. Interferon-alpha targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 2010, 38, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Desterke, C.; Bilhou-Nabéra, C.; Guerton, B.; Martinaud, C.; Tonetti, C.; Clay, D.; Guglielmelli, P.; Vannucchi, A.; Bordessoule, D.; Hasselbalch, H.; et al. FLT3-mediated p38-MAPK activation participates in the control of megakaryopoiesis in primary myelofibrosis. Cancer Res. 2011, 71, 2901–2915. [Google Scholar] [CrossRef] [Green Version]
- Srour, S.A.; Devesa, S.S.; Morton, L.M.; Check, D.P.; Curtis, R.E.; Linet, M.S.; Dores, G.M. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br. J. Haematol. 2016, 174, 382–396. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [Green Version]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, A.G. Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediators Inflamm. 2015, 2015, 606819. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stetka, J.; Gursky, J.; Liñan Velasquez, J.; Mojzikova, R.; Vyhlidalova, P.; Vrablova, L.; Bartek, J.; Divoky, V. Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes. Cancers 2020, 12, 903. https://doi.org/10.3390/cancers12040903
Stetka J, Gursky J, Liñan Velasquez J, Mojzikova R, Vyhlidalova P, Vrablova L, Bartek J, Divoky V. Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes. Cancers. 2020; 12(4):903. https://doi.org/10.3390/cancers12040903
Chicago/Turabian StyleStetka, Jan, Jan Gursky, Julie Liñan Velasquez, Renata Mojzikova, Pavla Vyhlidalova, Lucia Vrablova, Jiri Bartek, and Vladimir Divoky. 2020. "Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes" Cancers 12, no. 4: 903. https://doi.org/10.3390/cancers12040903
APA StyleStetka, J., Gursky, J., Liñan Velasquez, J., Mojzikova, R., Vyhlidalova, P., Vrablova, L., Bartek, J., & Divoky, V. (2020). Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes. Cancers, 12(4), 903. https://doi.org/10.3390/cancers12040903