ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms


Abstract
:1. Introduction
2. Results
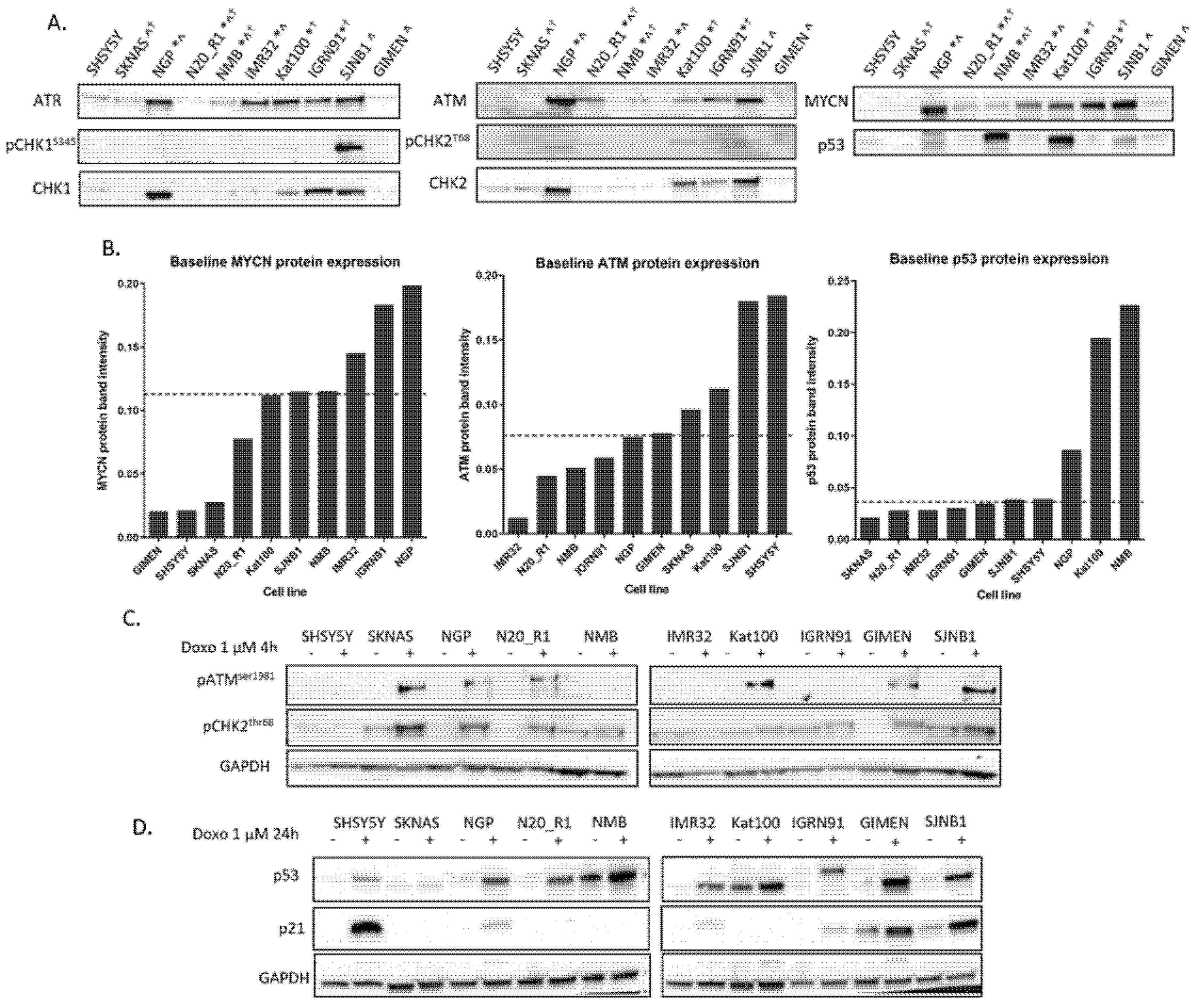
2.1. DDR Protein Expression in NB Cell Lines
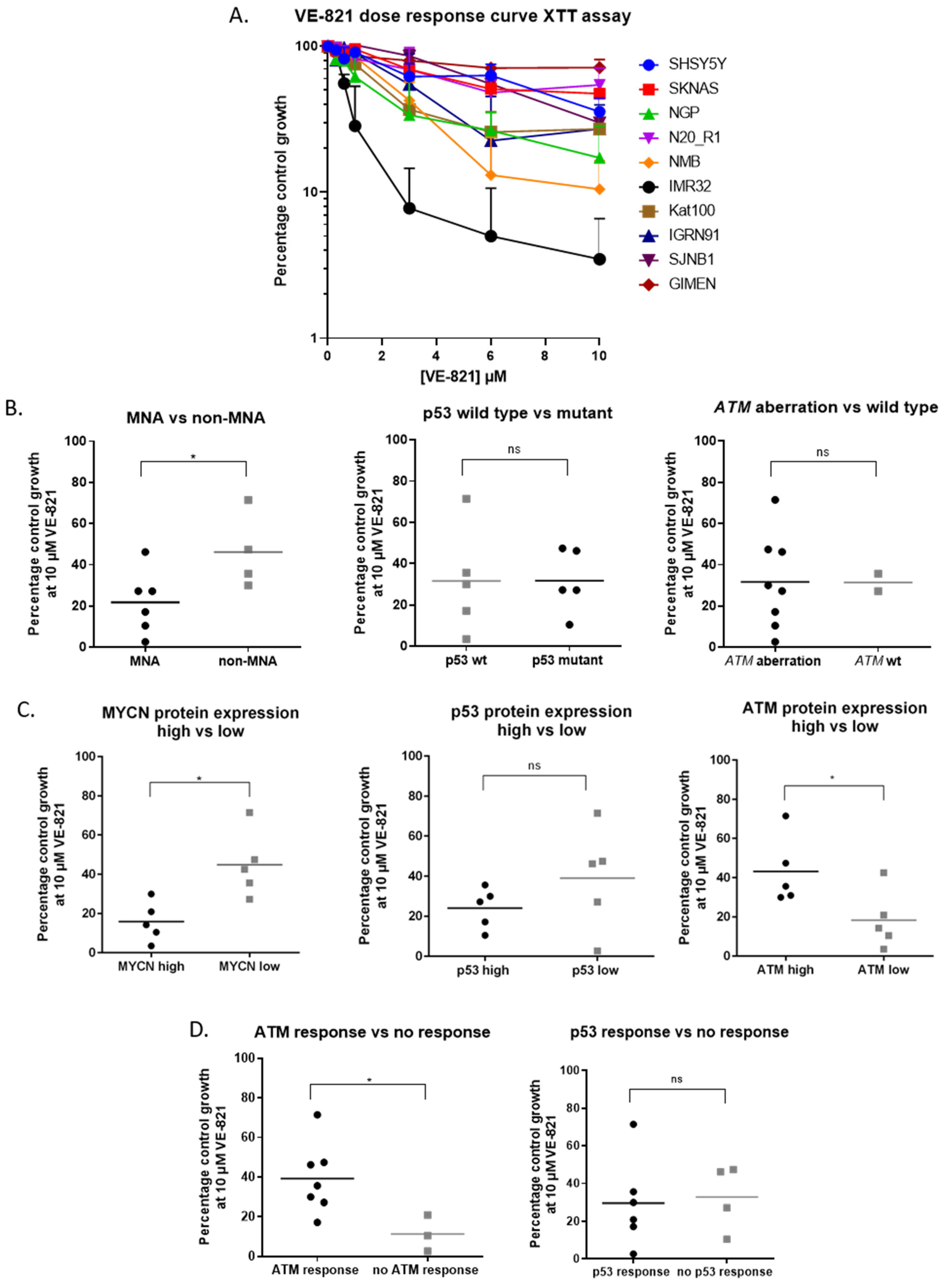
2.2. MYCN Amplification and Low ATM Expression are Determinants of Sensitivity to ATR Inhibition in NB Cell Lines
2.3. MYCN Amplification and Low ATM Expression Are Not Significant Determinants of Sensitivity to PARP Inhibition in NB Cell Lines
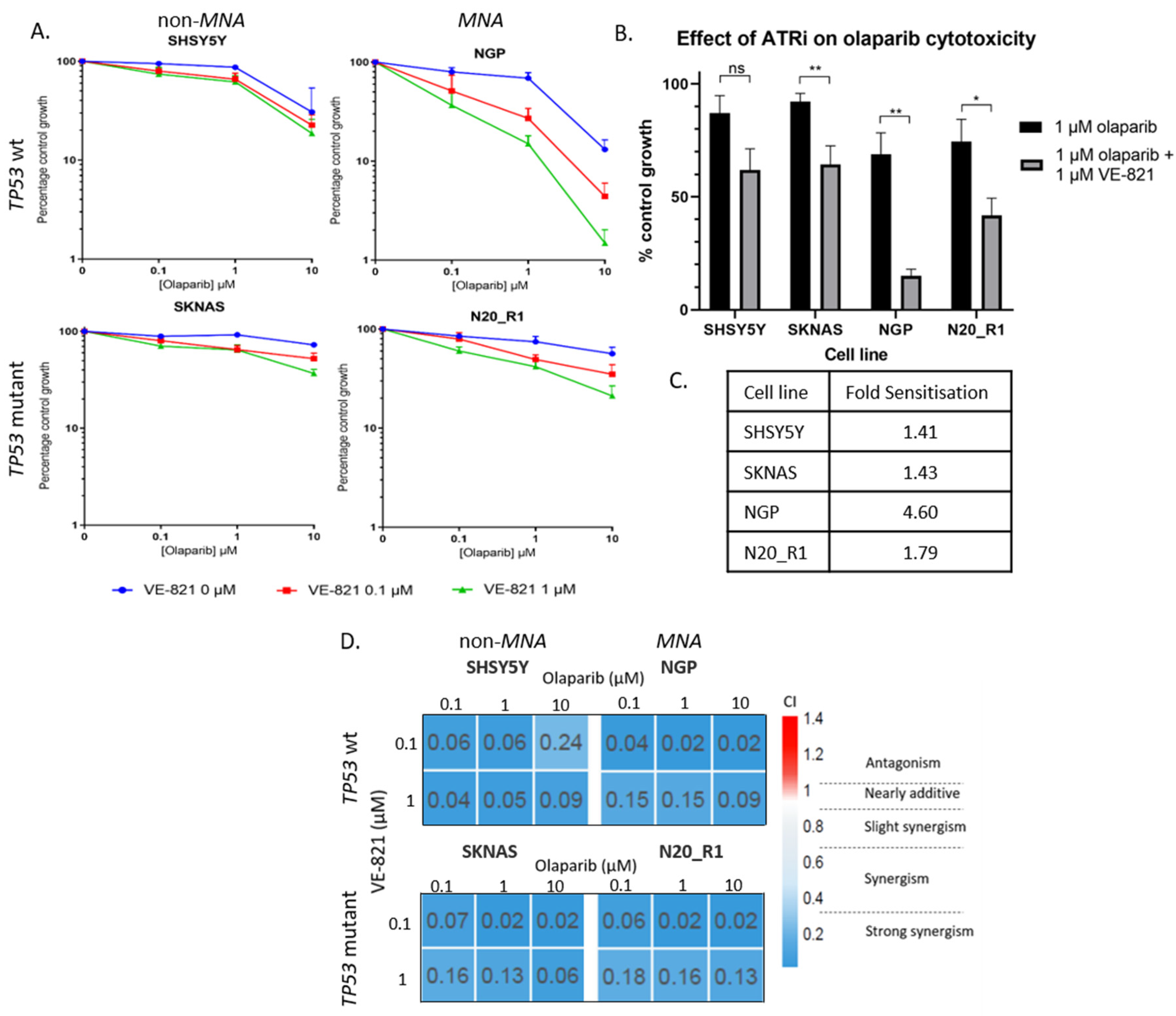
2.4. PARP and ATR Inhibition Synergistically Inhibit Cell Growth
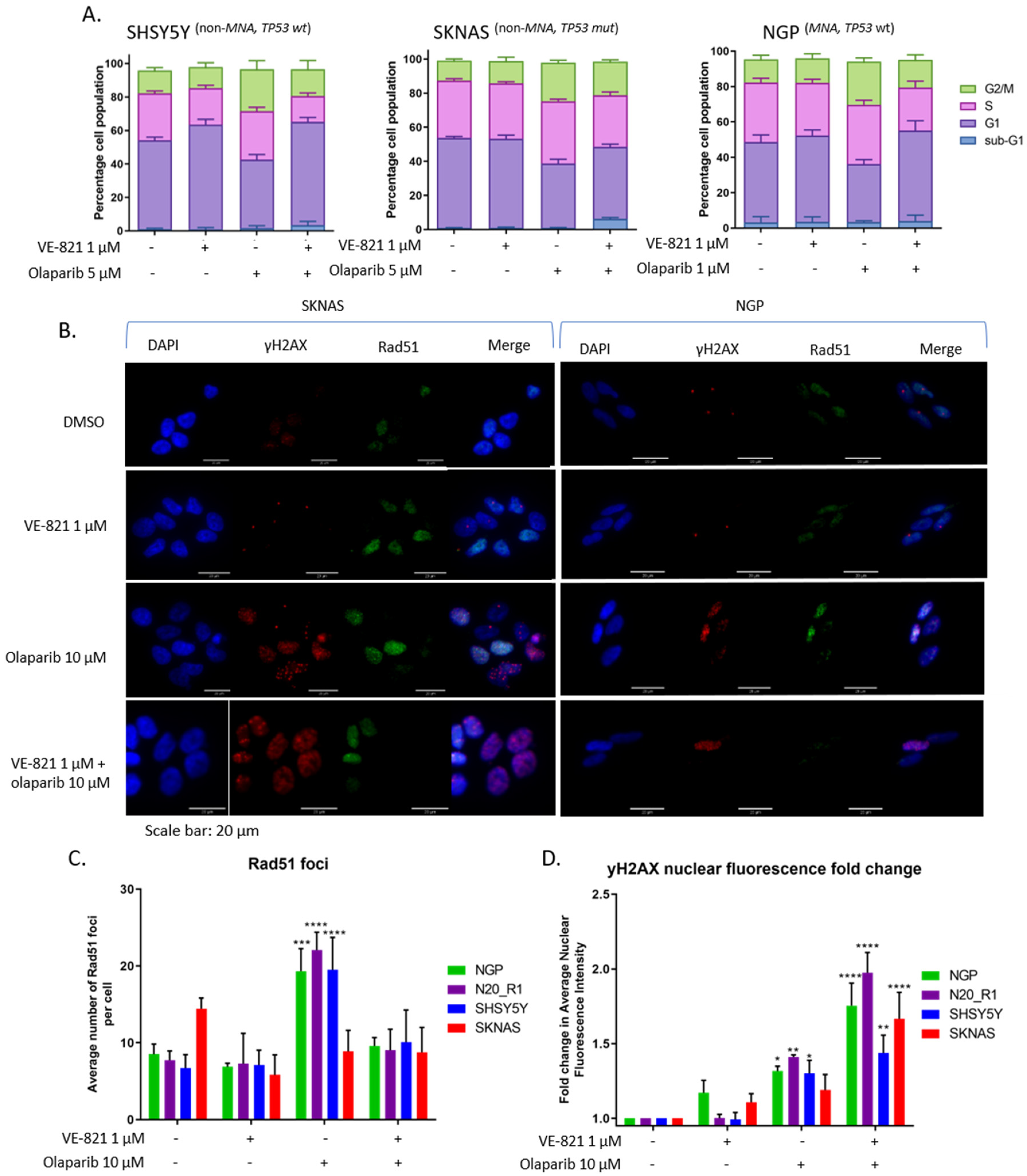
2.5. ATR Inhibition Increases Replication Stress Caused by Olaparib by Blocking S and G2 Cell Cycle Arrest and Reducing Homologous Recombination Repair
3. Discussion
4. Materials and Methods
4.1. Chemicals and Cell Lines
4.2. Growth Inhibition Assay
4.3. Protein Extraction and Western Blotting
4.4. Cell Cycle Analysis
4.5. Immunofluorescence
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yazinski, S.A.; Zou, L. Functions, regulation, and therapeutic implications of the atr checkpoint pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Lecona, E.; Fernandez-Capetillo, O. Targeting atr in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. Atm, atr, and DNA-pk: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.; Cortez, D.; Cimprich, K. The essential kinase atr: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. Atr inhibition induces synthetic lethality and overcomes chemoresistance in tp53- or atm-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Goetz, M.P.; et al. Atr inhibition is a promising radiosensitizing strategy for triple-negative breast cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef] [Green Version]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of atm-or p53-deficient cancer cells through inhibition of atr. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Zou, L. Molecular pathways: Targeting atr in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.M.; Lindsay, H.D. DNA replication stress and cancer: Cause or cure? Future Oncol. 2016, 12, 221–237. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Deckbar, D.; Birraux, J.; Krempler, A.; Tchouandong, L.; Beucher, A.; Walker, S.; Stiff, T.; Jeggo, P.; Löbrich, M. Chromosome breakage after g2 checkpoint release. J. Cell Biol. 2007, 176, 749–755. [Google Scholar] [CrossRef]
- Stolz, A.; Bastians, H. Therapeutic s and g2 checkpoint override causes centromere fragmentation in mitosis. Cell Cycle (Georget. Tex.) 2013, 12, 1826–1827. [Google Scholar] [CrossRef]
- Russell, K.J.; Wiens, L.W.; Demers, G.W.; Galloway, D.A.; Plon, S.E.; Groudine, M. Abrogation of the g2 checkpoint results in differential radiosensitization of g1 checkpoint-deficient and g1 checkpoint-competent cells. Cancer Res. 1995, 55, 1639–1642. [Google Scholar] [PubMed]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.G.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C. Childhood Cancer in Britain: Incidence, Survival, Mortality; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D. The international neuroblastoma risk group (inrg) classification system: An inrg task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.M.; Hogarty, M.; COG Neuroblastoma Committee. Children’s oncology group’s 2013 blueprint for research: Neuroblastoma. Pediatric Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Basta, N.O.; Halliday, G.C.; Makin, G.; Birch, J.; Feltbower, R.; Bown, N.; Elliott, M.; Moreno, L.; Barone, G.; Pearson, A.D.J.; et al. Factors associated with recurrence and survival length following relapse in patients with neuroblastoma. Br. J. Cancer 2016, 115, 1048. [Google Scholar] [CrossRef] [Green Version]
- Mlakar, V.; Mlakar, S.J.; Lopez, G.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. 11q deletion in neuroblastoma: A review of biological and clinical implications. Mol. Cancer 2017, 16, 114. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat Rev Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Carr-Wilkinson, J.; O’Toole, K.; Wood, K.M.; Challen, C.C.; Baker, A.G.; Board, J.R.; Evans, L.; Cole, M.; Cheung, N.-K.V.; Boos, J.; et al. High frequency of p53/mdm2/p14arf pathway abnormalities in relapsed neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1108–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.; Premkumar, R.; Carr, J.; Lu, X.; Lovat, P.E.; Kees, U.R.; Lunec, J.; Tweddle, D. The role of mycn in the failure of mycn amplified neuroblastoma cell lines to g1 arrest after DNA damage. Cell Cycle 2006, 5, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Kryh, H.; Nethander, M.; Sjöberg, R.-M.; Träger, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, J.; Bell, E.; Pearson, A.D.J.; Kees, U.R.; Beris, H.; Lunec, J.; Tweddle, D.A. Increased frequency of aberrations in the p53/mdm2/p14arf pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006, 66, 2138–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. Parp is activated at stalled forks to mediate mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Daniel, R.A.; Rozanska, A.L.; Thomas, H.D.; Mulligan, E.A.; Drew, Y.; Castelbuono, D.J.; Hostomsky, Z.; Plummer, E.R.; Boddy, A.V.; Tweddle, D.A.; et al. Inhibition of poly(adp-ribose) polymerase-1 enhances temozolomide and topotecan activity against childhood neuroblastoma. Clin. Cancer Res. 2009, 15, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Sanmartín, E.; Muñoz, L.; Piqueras, M.; Sirerol, J.A.; Berlanga, P.; Cañete, A.; Castel, V.; de Mora, J.F. Deletion of 11q in neuroblastomas drives sensitivity to parp inhibition. Clin. Cancer Res. 2017, 23, 6875–6887. [Google Scholar] [CrossRef] [Green Version]
- Nile, D.L.; Rae, C.; Hyndman, I.J.; Gaze, M.N.; Mairs, R.J. An evaluation in vitro of parp-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC Cancer 2016, 16, 621. [Google Scholar] [CrossRef] [Green Version]
- Norris, R.E.; Adamson, P.C.; Nguyen, V.T.; Fox, E. Preclinical evaluation of the parp inhibitor, olaparib, in combination with cytotoxic chemotherapy in pediatric solid tumors. Pediatric Blood Cancer 2014, 61, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahún-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F. Parp inhibitors enhance replication stress and cause mitotic catastrophe in mycn-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Yoshida, M.; Nemoto, Y.; Tamaichi, H.; Tsuchida, R.; Seki, M.; Uryu, K.; Nishii, R.; Miyamoto, S.; Saito, M. Loss of DNA damage response in neuroblastoma and utility of a parp inhibitor. J. Natl. Cancer Inst. 2017, 109, djx062. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.A.; Spengler, B.A.; Biedler, J.L. Coordinate morphological and biochemical interconversion of human neuroblastoma cells2. JNCI J. Natl. Cancer Inst. 1983, 71, 741–747. [Google Scholar] [PubMed]
- Goldschneider, D.; Horvilleur, E.; Plassa, L.-F.; Guillaud-Bataille, M.; Million, K.; Wittmer-Dupret, E.; Danglot, G.; de Thé, H.; Bénard, J.; May, E.; et al. Expression of c-terminal deleted p53 isoforms in neuroblastoma. Nucleic Acids Res. 2006, 34, 5603–5612. [Google Scholar] [CrossRef] [Green Version]
- Van Roy, N.; Van Limbergen, H.; Vandesompele, J.; Van Gele, M.; Poppe, B.; Salwen, H.; Laureys, G.; Manoel, N.; De Paepe, A.; Speleman, F. Combined m-fish and cgh analysis allows comprehensive description of genetic alterations in neuroblastoma cell lines. GenesChromosomes Cancer 2001, 32, 126–135. [Google Scholar] [CrossRef]
- Drummond, C.J.; Esfandiari, A.; Liu, J.; Lu, X.; Hutton, C.; Jackson, J.; Bennaceur, K.; Xu, Q.; Makimanejavali, A.R.; Bello, F.D.; et al. Tp53 mutant mdm2-amplified cell lines selected for resistance to mdm2-p53 binding antagonists retain sensitivity to ionizing radiation. Oncotarget 2016, 7, 46203–46218. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.C.; Chen, C.Y.; Lansing, T.J.; Gilmer, T.M.; Kastan, M.B. The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. Am. J. Pathol. 1996, 148, 1381–1385. [Google Scholar]
- Tumilowicz, J.J.; Nichols, W.W.; Cholon, J.J.; Greene, A.E. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970, 30, 2110–2118. [Google Scholar]
- Mandriota, S.J.; Valentijn, L.J.; Lesne, L.; Betts, D.R.; Marino, D.; Boudal-Khoshbeen, M.; London, W.B.; Rougemont, A.-L.; Attiyeh, E.F.; Maris, J.M. Ataxia-telangiectasia mutated (atm) silencing promotes neuroblastoma progression through a mycn independent mechanism. Oncotarget 2015, 6, 18558. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Haber, M.; Flemming, C.; Marshall, G.M.; Lock, R.B.; MacKenzie, K.L.; Gurova, K.V.; Norris, M.D.; Gudkov, A.V. P53 determines multidrug sensitivity of childhood neuroblastoma. Cancer Res. 2007, 67, 10351–10360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, E.; Goldschneider, D.; Ferrandis, E.; Barrois, M.; Le Roux, G.; Leonce, S.; Douc-Rasy, S.; Bénard, J.; Raguénez, G. Mycn enhances p-gp/mdr1 gene expression in the human metastatic neuroblastoma igr-n-91 model. Am. J. Pathol. 2003, 163, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Flahaut, M.; Mühlethaler-Mottet, A.; Martinet, D.; Fattet, S.; Bourloud, K.B.; Auderset, K.; Meier, R.; Schmutz, N.B.; Delattre, O.; Joseph, J.-M.; et al. Molecular cytogenetic characterization of doxorubicin-resistant neuroblastoma cell lines: Evidence that acquired multidrug resistance results from a unique large amplification of the 7q21 region. GenesChromosomes Cancer 2006, 45, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication catastrophe: When a checkpoint fails because of exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-pk, atm and atr in rpa phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [Green Version]
- Middleton, F.K.; Pollard, J.R.; Curtin, N.J. The impact of p53 dysfunction in atr inhibitor cytotoxicity and chemo- and radiosensitisation. Cancers 2018, 10, 275. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of parp1 and parp2 by clinical parp inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Levine, D.A. New insights into parp inhibitors’ effect on cell cycle and homology-directed DNA damage repair. Mol. Cancer Ther. 2014, 13, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. Parp inhibitors: Synthetic lethality in the clinic. Science (New York N.Y.) 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(adp-ribose) polymerase inhibitors. Clin. Cancer Res. 2010, 16, 2344–2351. [Google Scholar] [CrossRef] [Green Version]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel atr inhibitor, nu6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. Atr inhibition disrupts rewired homologous recombination and fork protection pathways in parp inhibitor-resistant brca-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of atr. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; D’Artista, L.; Schleker, T.; Guerra, C.; Garcia, E. Exploiting oncogene-induced replicative stress for the selective killing of myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies atr inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Petroni, M.; Sardina, F.; Infante, P.; Bartolazzi, A.; Locatelli, E.; Fabretti, F.; Di Giulio, S.; Capalbo, C.; Cardinali, B.; Coppa, A.; et al. Mre11 inhibition highlights a replication stress-dependent vulnerability of mycn-driven tumors. Cell Death Dis. 2018, 9, 895. [Google Scholar] [CrossRef]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; McCormick, A.; Edmondson, R.; May, F.E.B.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent atr inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.-P.; George, J.; Nowak, M.; Leeser, U.; Büttner, R.; Perner, S.; Peifer, M.; et al. Atm deficiency is associated with sensitivity to parp1- and atr inhibitors in lung adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Im, S.-A.; Jang, H.; Kim, S.; Lee, M.; Kim, D.K.; Yang, Y.; Kim, H.-J.; Lee, K.-H.; Kim, J.W.; et al. Azd6738, a novel oral inhibitor of atr, induces synthetic lethality with atm deficiency in gastric cancer cells. Mol. Cancer Ther. 2017, 16, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Du, L.; Nagabayashi, G.; Seeger, R.C.; Gatti, R.A. Atm is down-regulated by n-myc-regulated microrna-421. Proc. Natl. Acad. Sci. USA 2010, 107, 1506–1511. [Google Scholar] [CrossRef] [Green Version]
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A synthetic lethal screen identifies DNA repair pathways that sensitize cancer cells to combined atr inhibition and cisplatin treatments. PLoS ONE 2015, 10, e0125482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the atr/chk1 axis with parp inhibition results in tumor regression in brca-mutant ovarian cancer models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y. Resistance to parp inhibitors by slfn11 inactivation can be overcome by atr inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.L.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of homologous recombination and the checkpoint by atr. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Bianco, J.N.; Bergoglio, V.; Lin, Y.-L.; Pillaire, M.-J.; Schmitz, A.-L.; Gilhodes, J.; Lusque, A.; Mazières, J.; Lacroix-Triki, M.; Roumeliotis, T.I.; et al. Overexpression of claspin and timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat. Commun. 2019, 10, 910. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Gao, Y.; Mutter-Rottmayer, L.; Zlatanou, A.; Durando, M.; Ding, W.; Wyatt, D.; Ramsden, D.; Tanoue, Y.; Tateishi, S.; et al. DNA repair factor rad18 and DNA polymerase polκ confer tolerance of oncogenic DNA replication stress. J. Cell Biol. 2017, 216, 3097–3115. [Google Scholar] [CrossRef] [Green Version]
- Kurashima, K.; Sekimoto, T.; Oda, T.; Kawabata, T.; Hanaoka, F.; Yamashita, T. Polη, a y-family translesion synthesis polymerase, promotes cellular tolerance of myc-induced replication stress. J. Cell Sci. 2018, 131, jcs212183. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | MYCN Status | 11q Status (Genes Deleted) | p53 Status | Reference |
|---|---|---|---|---|
| SHSY5Y | Non-amp | No deletion | WT | [34] |
| SKNAS | Non-amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | Mutant Deletion of intron9/exon 10 | [35,36] |
| NGP | Amp | Deletion (ATM, CHEK1, H2AFX) | WT | [36] |
| N20_R1 | Amp | Deletion ** (ATM, CHEK1, H2AFX) | Mutant P98H P152T | [37] |
| NMB * | Amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | Mutant G245S | [36,38] |
| IMR32 | Amp | Deletion (ATM, CHEK1, H2AFX) ATM mutant V2716A | WT | [39,40] |
| IMR32/Kat100 (Kat100) | Amp | Unknown | Mutant C135F | [41] |
| IGRN91 | Amp | No deletion | Mutant Duplication of exons 7–9 | [42,43] |
| SJNB1 * | Non-amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | WT | [36] |
| GIMEN | Non-amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | WT | [36] |
| Cell Line | MYCN Status | 11q Status (Genes Deleted) | p53 Status | VE-821 GI50 (μM) | VE-821 LC50 (μM) | Olaparib GI50 (μM) | Olaparib LC50 (μM) |
|---|---|---|---|---|---|---|---|
| SHSY5Y | Non-amp | No deletion | WT | 7.11 | 1.54 | 5.38 | 1.40 |
| SKNAS | Non-amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | Mut | >20 | 0.81 | >30 | 1.31 |
| NGP | Amp | Deletion (ATM, CHEK1, H2AFX) | WT | 1.62 | 0.93 | 1.35 | 1.20 |
| N20_R1 | Amp | Deletion (ATM, CHEK1, H2AFX) | Mut | 8.29 | 0.93 | 1.64 | 0.68 |
| NMB | Amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | Mut | 2.36 | 1.91 | 3.88 | 0.92 |
| IMR32 | Amp | Deletion (ATM, CHEK1, H2AFX) ATM mutant V2716A | WT | 0.66 | 0.90 | 1.81 | 0.63 |
| Kat100 | Amp | Unknown | Mut | 1.88 | 1.50 | >30 | 1.55 |
| IGRN91 | Amp | No deletion | Mut | 3.04 | 1.03 | >30 | 0.74 |
| SJNB1 | Non-amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | WT | 6.30 | 1.99 | 4.70 | 0.75 |
| GIMEN | Non-amp | Deletion (MRE11, ATM, CHEK1, H2AFX) | WT | >20 | 0.87 | 12.25 | 0.37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Southgate, H.E.D.; Chen, L.; Tweddle, D.A.; Curtin, N.J. ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms. Cancers 2020, 12, 1095. https://doi.org/10.3390/cancers12051095
Southgate HED, Chen L, Tweddle DA, Curtin NJ. ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms. Cancers. 2020; 12(5):1095. https://doi.org/10.3390/cancers12051095
Chicago/Turabian StyleSouthgate, Harriet E. D., Lindi Chen, Deborah A. Tweddle, and Nicola J. Curtin. 2020. "ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms" Cancers 12, no. 5: 1095. https://doi.org/10.3390/cancers12051095
APA StyleSouthgate, H. E. D., Chen, L., Tweddle, D. A., & Curtin, N. J. (2020). ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms. Cancers, 12(5), 1095. https://doi.org/10.3390/cancers12051095