The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer


 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
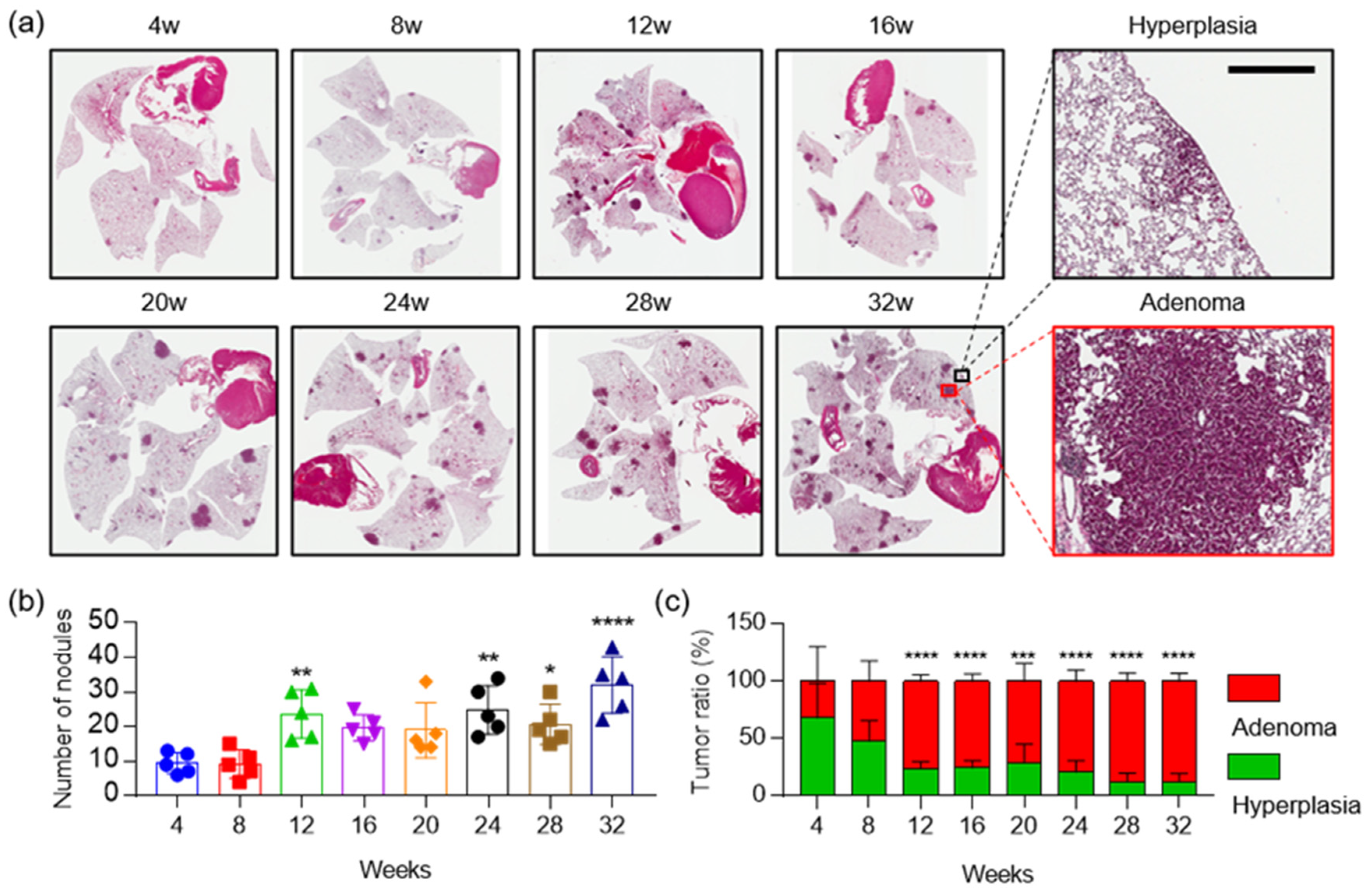
2.1. Aldh1l1 Expression is Associated with KRAS Mutation in Lung Cancer Cell Lines
2.2. KRAS Response Region in the ALDH1L1 Promoter
2.3. Generation of Aldh1l1-Deficient Mice
2.4. The Combination of Aldh1l1 Deficiency and Phenformin Treatment Suppresses KRAS-Driven Lung Tumorigenesis
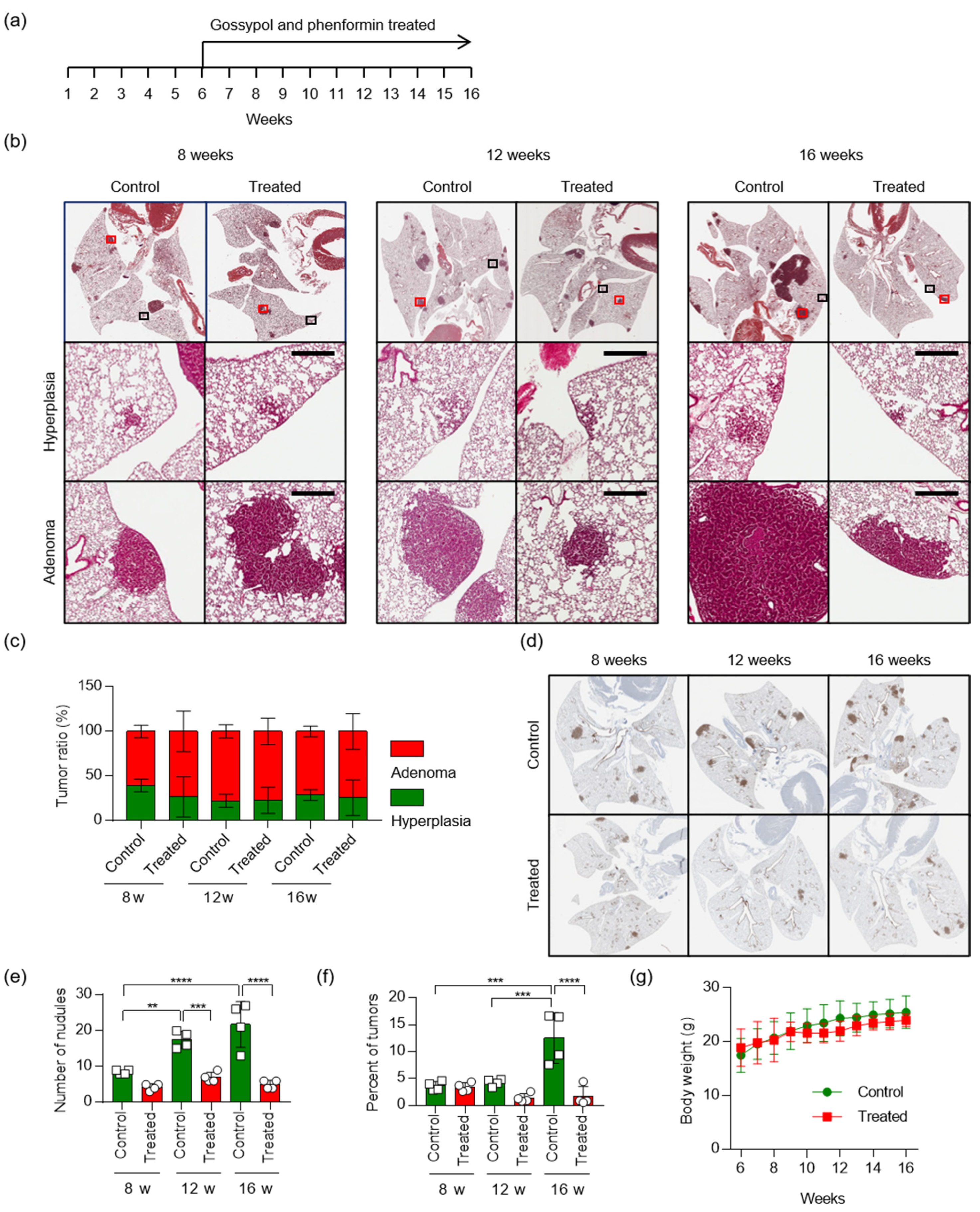
2.5. Treatment with Gossypol and Phenformin Suppresses KRAS-Driven Lung Tumorigenesis
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies and Reagents
4.3. Immunoblotting
4.4. ALDH1L1 Promoter Assay
4.5. Spontaneous Lung Cancer Model
4.6. Automated Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roman, M.; Baraibar, I.; Lopez, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer 2018, 17, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlaender, A.; Drilon, A.; Weiss, G.J.; Banna, G.L.; Addeo, A. KRAS as a druggable target in NSCLC: Rising like a phoenix after decades of development failures. Cancer Treat. Rev. 2020, 85, 101978. [Google Scholar] [CrossRef] [PubMed]
- Rissmann, R.; Hessel, M.H.; Cohen, A.F. Vemurafenib/dabrafenib and trametinib. Br. J. Clin. Pharmacol. 2015, 80, 765–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [Green Version]
- Dias Carvalho, P.; Guimaraes, C.F.; Cardoso, A.P.; Mendonca, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wan, L.; Geng, J.; Wu, C.L.; Bai, X. Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 2012, 7, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, S.H.; Lee, J.S.; Nam, B.; Seong, T.W.; Son, J.; Jang, H.; Hong, K.M.; Lee, C.; Kim, S.Y. Aldehyde dehydrogenase inhibition combined with phenformin treatment reversed NSCLC through ATP depletion. Oncotarget 2016, 7, 49397–49410. [Google Scholar] [CrossRef]
- Cook, R.J.; Lloyd, R.S.; Wagner, C. Isolation and characterization of cDNA clones for rat liver 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 1991, 266, 4965–4973. [Google Scholar] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Anguera, M.C.; Field, M.S.; Perry, C.; Ghandour, H.; Chiang, E.P.; Selhub, J.; Shane, B.; Stover, P.J. Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 2006, 281, 18335–18342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Boyer, J.L.; Herbst, R.S. A RAS renaissance: Emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 3921–3930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, O.A.; Sandi, M.J.; Burston, H.E.; Brown, K.R.; Rottapel, R. An oncogenic KRAS transcription program activates the RHOGEF ARHGEF2 to mediate transformed phenotypes in pancreatic cancer. Oncotarget 2017, 8, 4484–4500. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Barillo, J.L.; da Silva, C.T., Jr.; Silva, P.S.; de Souza, J.B.S.; Kanaan, S.; Xavier, A.R.; de Araujo, E.G. Increased cytokeratin 19 fragment levels are positively correlated with adenosine deaminase activity in malignant pleural effusions from adenocarcinomas. Dis. Markers 2018, 2018, 2609767. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupenko, S.A. FDH: An aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem. Biol. Interact. 2009, 178, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupenko, S.A.; Wagner, C.; Cook, R.J. Expression, purification, and properties of the aldehyde dehydrogenase homologous carboxyl-terminal domain of rat 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 1997, 272, 10266–10272. [Google Scholar] [CrossRef] [Green Version]
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 1969, 115, 609–619. [Google Scholar] [CrossRef]
- Lin, S.J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Krupenko, S.A.; Krupenko, N.I. ALDH1L1 and ALDH1L2 folate regulatory enzymes in cancer. Adv. Exp. Med. Biol. 2018, 1032, 127–143. [Google Scholar] [PubMed]
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. 1998, 274, F315–F327. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Jeon, Y.; Kang, J.H.; Jang, H.; Lee, H.; Kim, S.-Y. The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers 2020, 12, 1382. https://doi.org/10.3390/cancers12061382
Lee S-H, Jeon Y, Kang JH, Jang H, Lee H, Kim S-Y. The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers. 2020; 12(6):1382. https://doi.org/10.3390/cancers12061382
Chicago/Turabian StyleLee, Seon-Hyeong, Yoon Jeon, Joon Hee Kang, Hyonchol Jang, Ho Lee, and Soo-Youl Kim. 2020. "The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer" Cancers 12, no. 6: 1382. https://doi.org/10.3390/cancers12061382
APA StyleLee, S. -H., Jeon, Y., Kang, J. H., Jang, H., Lee, H., & Kim, S. -Y. (2020). The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers, 12(6), 1382. https://doi.org/10.3390/cancers12061382