Characterization of a Myeloid Activation Signature That Correlates with Survival in Melanoma Patients

 , , and
, , and
Abstract
:1. Introduction
2. Results
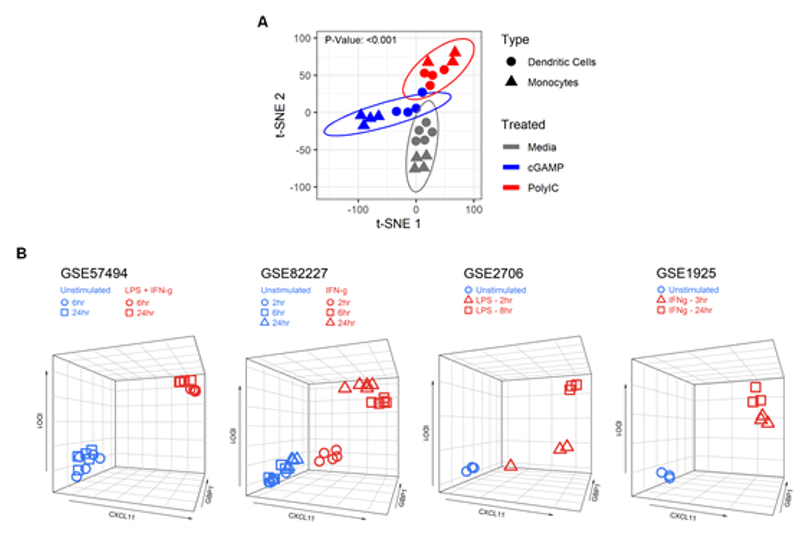
2.1. Identification of a Myeloid Cell Activation Signature
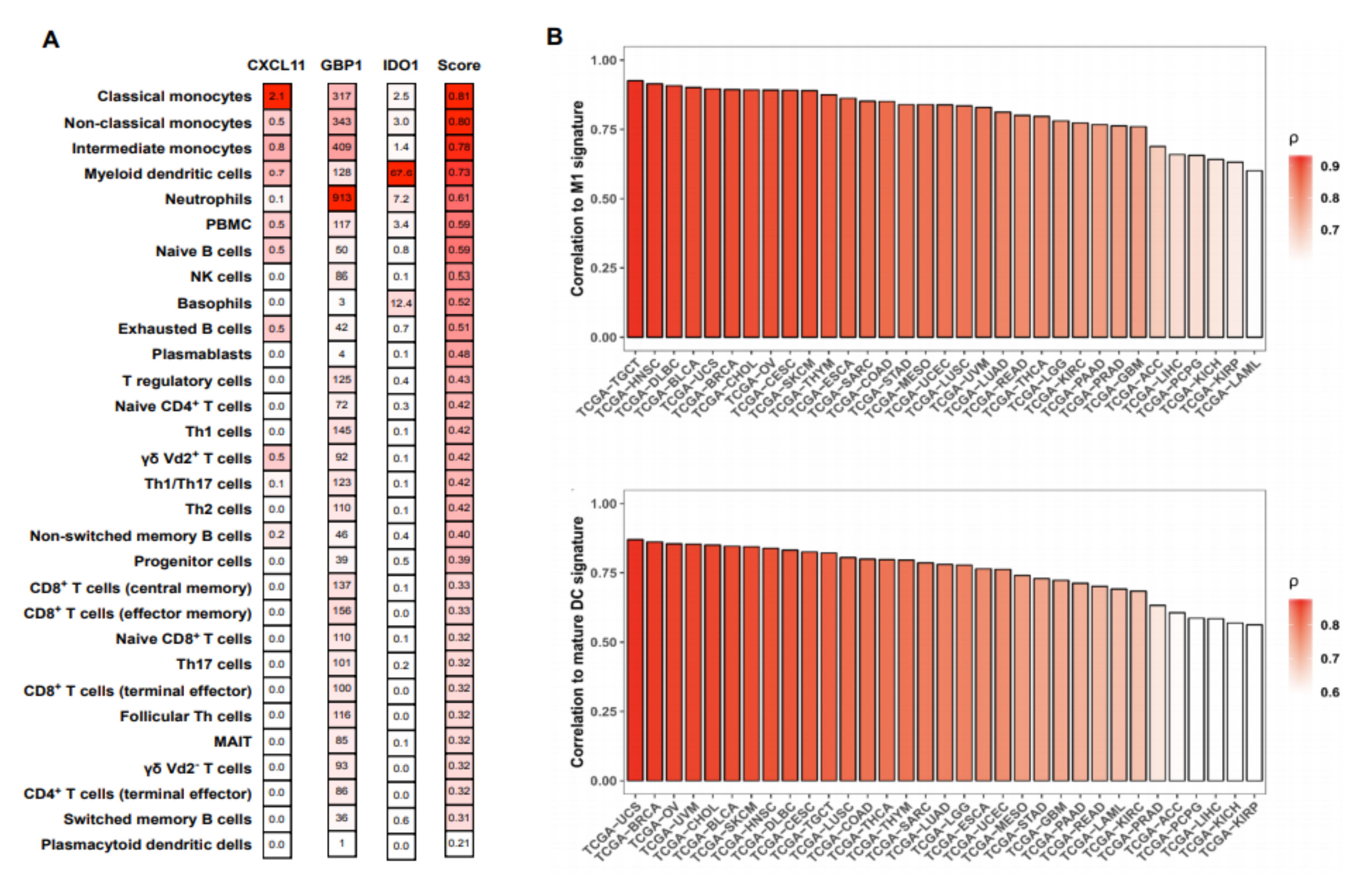
2.2. The MA Gene Signature is Primarily Expressed in Activated Cells of the Myeloid Lineage
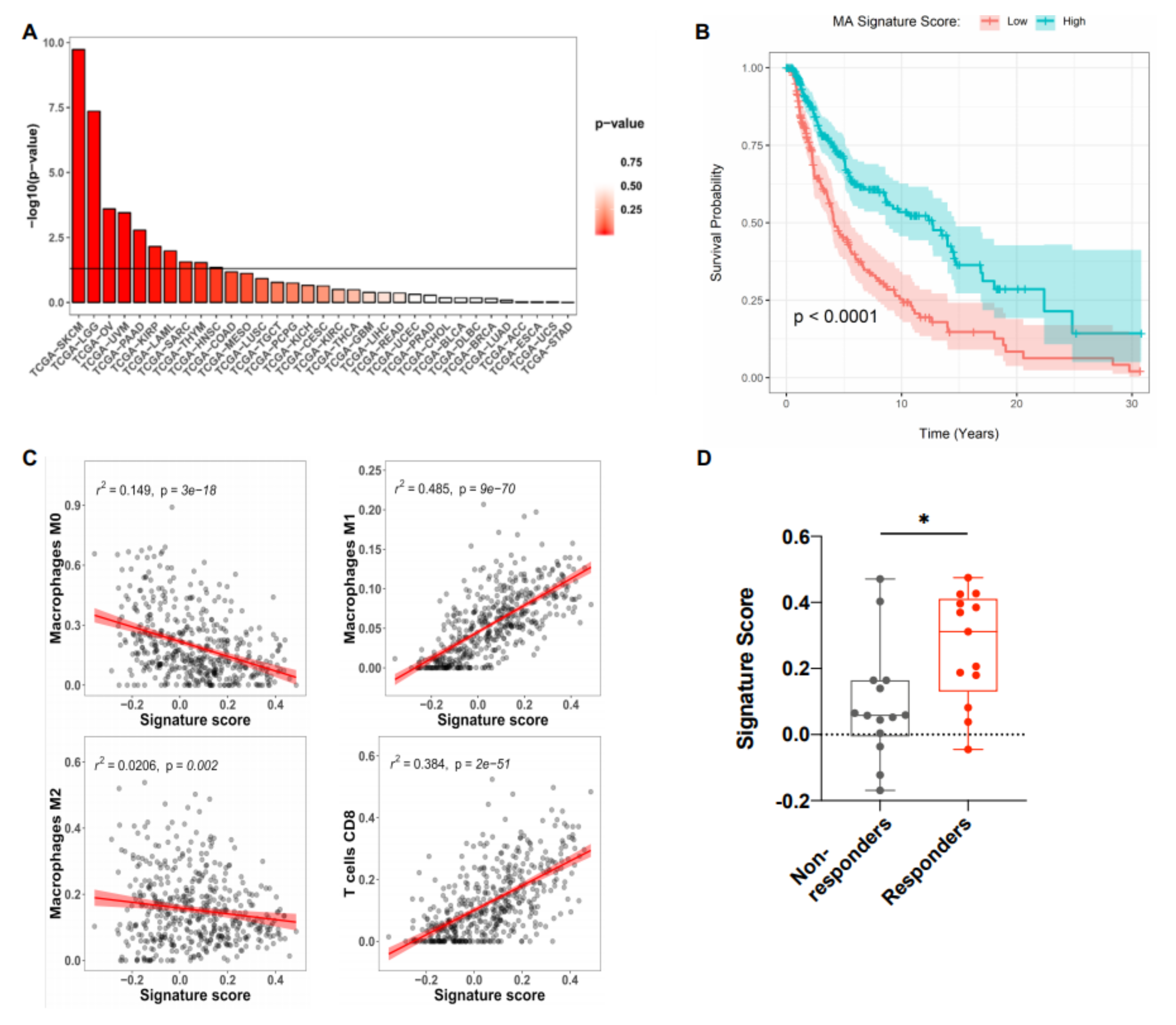
2.3. The MA Gene Signature Correlated with Improved Overall Survival in Melanoma Patients
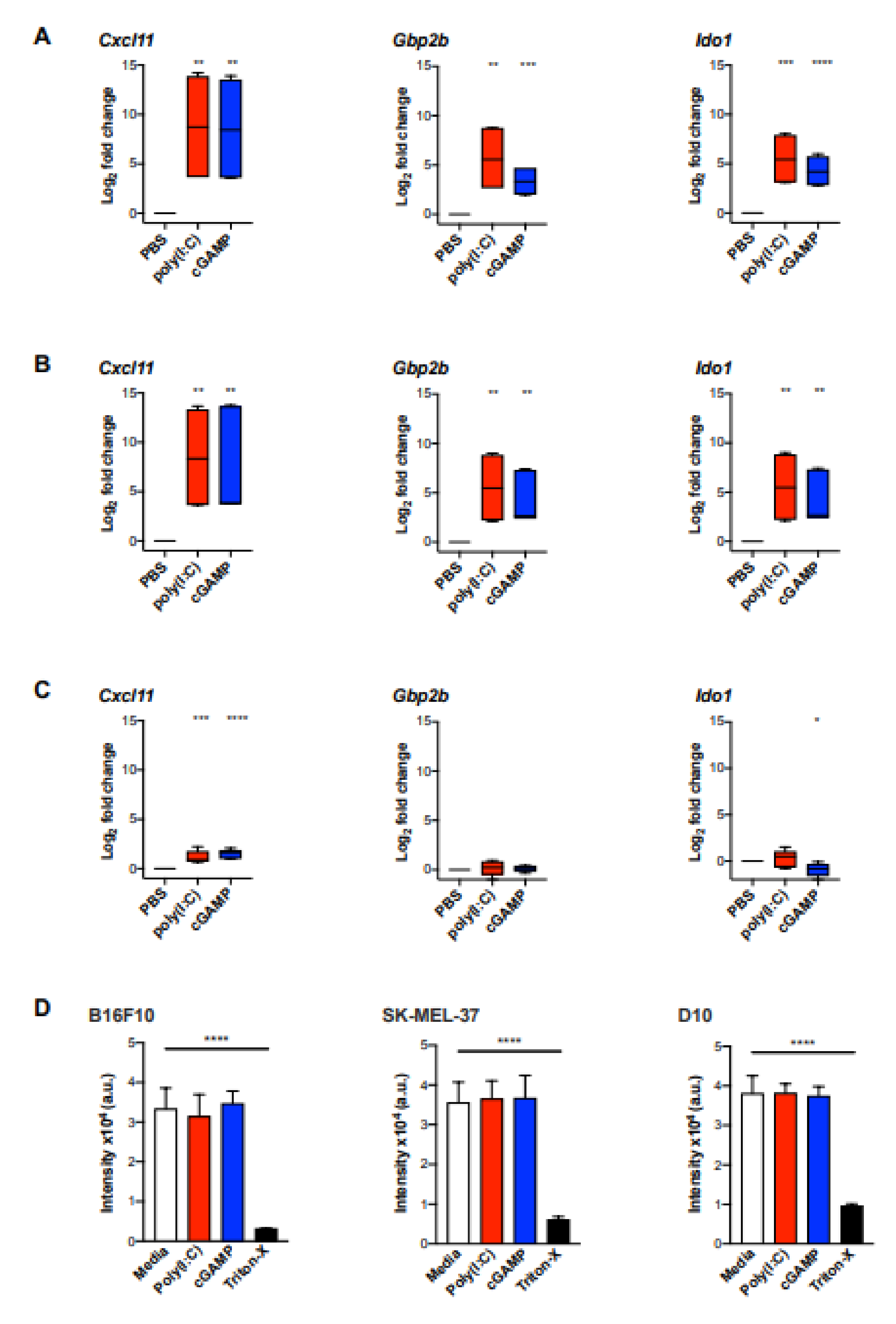
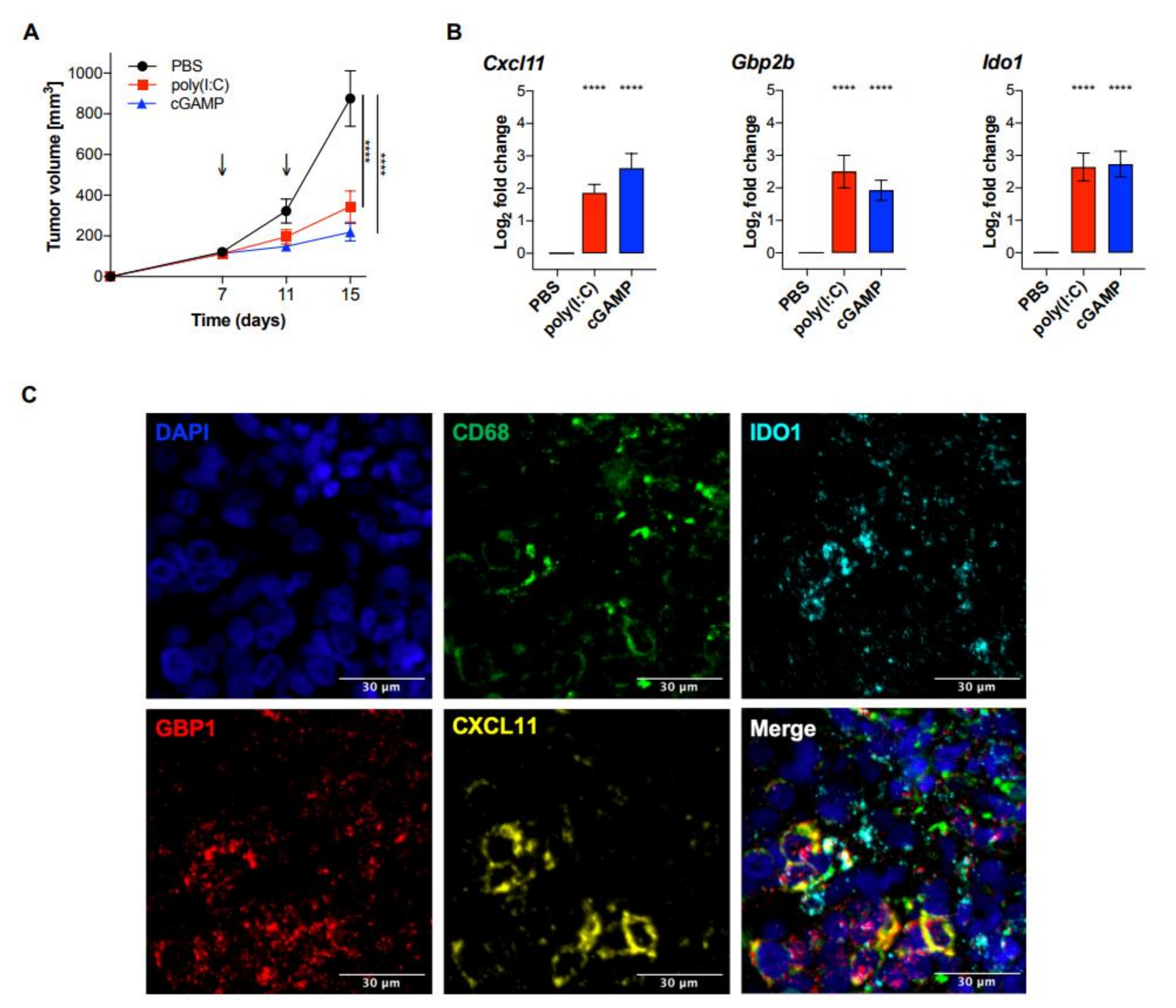
2.4. Validation of the MA Signature in Murine Myeloid Cells and In Vivo Murine Melanoma
3. Discussion
4. Materials and Methods
4.1. Tissue Culture
4.2. Human Monocytes and DC
4.3. NanoString mRNA Profiling
4.4. BMDM Differentiation and Maturation for In Vitro Studies
4.5. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.6. AlamarBlue Assay
4.7. Mice Tumor Inoculation and In Vivo Studies
4.8. RNA In Situ Hybridization (RNAscope)
4.9. TCGA Data Analysis
4.10. Analysis of GEO Datasets
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 3, 55–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer Immunotherapy: Beyond Checkpoint Blockade. Annu. Rev. Cancer Biol. 2018, 168, 707–723. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, R.M.; De Vlaeminck, Y.; Maebe, J.; Goyvaerts, C.; Breckpot, K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front. Immunol. 2018, 9, 1977. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef]
- De Nardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Kitamura, T. Macrophage targeting: Opening new possibilities for cancer immunotherapy. Immunology 2018, 155, 285–293. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aleynick, M.; Svensson-Arvelund, J.; Flowers, C.R.; Marabelle, A.; Brody, J.D. Pathogen Molecular Pattern Receptor Agonists: Treating Cancer by Mimicking Infection. Clin. Cancer Res. 2019, 25, 6283–6294. [Google Scholar] [CrossRef]
- Shekarian, T.; Valsesia-Wittmann, S.; Brody, J.; Michallet, M.C.; Depil, S.; Caux, C.; Marabelle, A. Pattern recognition receptors: Immune targets to enhance cancer immunotherapy. Ann. Oncol. 2017, 28, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Raza Zaidi, M. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Yi, H.Y.; Chrisophe, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Han, T.H.; Ren, J.; Saunders, S.; Wang, E.; Marincola, F.M.; Stroncek, D.F. Molecular signatures of maturing dendritic cells: Implications for testing the quality of dendritic cell therapies. J. Transl. Med. 2010, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.; Bastiani, M.A.D.; Parisi, M.M.; Guma, F.T.C.R.; Markoski, M.M.; Castro, M.A.A.; Kaplan, M.H.; Barbé-Tuana, F.M.; Klamt, F. Integrated Transcriptomics Establish Macrophage Polarization Signatures and have Potential Applications for Clinical Health and Disease. Sci. Rep. 2015, 5, 13351. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; Giraldo, N.A.; Dieu-Nosjean, M.C.; Sautès-Fridman, C.; Fridman, W.H. Cancer immune contexture and immunotherapy. Curr. Opin. Immunol. 2016, 39, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]
- Hoang, V.L.T.; Tom, L.N.; Quek, X.-C.; Tan, J.-M.; Payne, E.J.; Lin, L.L.; Sinnya, S.; Raphael, A.P.; Lambie, D.; Frazer, I.H.; et al. RNA-seq reveals more consistent reference genes for gene expression studies in human non-melanoma skin cancers. PeerJ 2017, 5, e3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Tretina, K.; Park, E.S.; Maminska, A.; MacMicking, J.D. Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J. Exp. Med. 2019, 216, 482–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Guo, H.; Yang, J.; Ji, Y.; Wu, C.; Xiaoli, C. Down-regulation of guanylate binding protein 1 causes mitochondrial dysfunction and cellular senescence in macrophages. Sci. Rep. 2018, 8, 1679. [Google Scholar] [CrossRef]
- Li, F.; Zhang, R.; Li, S.; Liu, J. IDO1: An important immunotherapy target in cancer treatment. Int. Immunopharmacol. 2017, 47, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Newton, R.C.; Friedman, S.M.; Scherle, P.A. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9, 938–952. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Chung, D.J.; Rossi, M.; Romano, E.; Ghith, J.; Yuan, J.; Munn, D.H.; Young, J.W. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood 2009, 114, 555–563. [Google Scholar] [CrossRef]
- Wang, X.F.; Wang, H.-S.; Wang, H.; Zhang, F.; Wang, K.-F.; Guo, Q.; Zhang, G.; Cai, S.-H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Smith, C.; DuHadaway, J.B.; Sutanto-Ward, E.; Prendergast, G.C.; Bravo-Nuevo, A.; Muller, A.J. IDO1 is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine 2016, 14, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schollbach, J.; Kircher, S.; Wiegering, A.; Seyfried, F.; Klein, I.; Rosenwald, A.; Germer, C.T.; Löb, S. Prognostic value of tumour-infiltrating CD8+ lymphocytes in rectal cancer after neoadjuvant chemoradiation: Is indoleamine-2,3-dioxygenase (IDO1) a friend or foe? Cancer Immunol. Immunother. 2019, 68, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Ladányi, A.; Kiss, J.; Somlai, B.; Gilde, K.; Fejős, Z.; Mohos, A.; Gaudi, I.; Tímár, J. Density of DC-LAMP + mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol. Immunother. 2007, 56, 1459–1469. [Google Scholar] [CrossRef]
- Truxova, I.; Kasikova, L.; Hensler, M.; Skapa, P.; Laco, J.; Pecen, L.; Belicova, L.; Praznovec, I.; Halaska, M.J.; Brtnicky, T.; et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J. Immunother. Cancer 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nature Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shime, H.; Matsumoto, M.; Oshiumi, H.; Tanaka, S.; Nakane, A.; Iwakura, Y.; Tahara, H.; Inoue, N.; Seya, T. Toll-like receptor 3 signaling converts tumorsupporting myeloid cells to tumoricidal effectors. Proc. Natl. Acad. Sci. USA 2012, 6, 2066–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuri, T.; Kosaka, A.; Ishibashi, K.; Kumai, T.; Hirata, Y.; Ohara, K.; Nagato, T.; Oikawa, K.; Aoki, N.; Harabuchi, Y.; et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol. Immunother. 2017, 66, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, N.; Torensma, R.; Tel, J. Deciphering the message broadcast by tumor-infiltrating dendritic cells. Am. J. Pathol. 2012, 181, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Kiss, M.; Van Gassen, S.; Movahedi, K.; Saeys, Y.; Laoui, D. Myeloid cell heterogeneity in cancer: Not a single cell alike. Cell. Immunol. 2018, 330, 188–201. [Google Scholar] [CrossRef]
- Lyons, Y.A.; Wu, S.Y.; Overwijk, W.W.; Baggerly, K.A.; Sood, A.K. Immune cell profiling in cancer: Molecular approaches to cell-specific identification. NPJ Precis. Oncol. 2017, 1, 26. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Finotello, F.; Trajanoski, Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol. Immunother. 2018, 67, 1031–1040. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, S.; We, J.; Zhang, L.; Ma, R.; Lu, J.; Zhu, J.; Zhong, W.D.; Jia, Z. GDCRNATools: An R/Bioconductor package for integrative analysis of lncRNA, miRNA and mRNA data in GDC. Bioinformatics 2018, 34, 2515–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Monocytes | DC | ||
|---|---|---|---|---|
| cGAMP | Poly(I:C) | cGAMP | Poly(I:C) | |
| CCL3 | 5.26 | 5.48 | 3.7 | 4.83 |
| CCL4 | 4.97 | 4.87 | 3.62 | 5.14 |
| CXCL10 | 9.10 | 3.89 | 10.69 | 7.40 |
| CXCL11 | 9.96 | 3.42 | 11.21 | 8.96 |
| GBP1 | 4.79 | 2.31 | 4.37 | 3.91 |
| IDO1 | 6.82 | 4.26 | 2.67 | 3.41 |
| IFNB1 | 17.73 | 6.95 | 16.71 | 15.18 |
| IL27 | 15.05 | 10.80 | 11.03 | 11.54 |
| IL-29 | 13.44 | 8.86 | 15.03 | 14.28 |
| TGFBR2 | −3.73 | −3.89 | −9.72 | −9.91 |
| Genes | RNA-seq | Microarray | ||
|---|---|---|---|---|
| GSE57494 | GSE82227 | GSE2706 | GSE1925 | |
| CCL3 | <0.001 | 1.000 | 0.003 | 0.137 |
| CCL4 | <0.001 | 1.000 | 0.001 | 0.027 |
| CXCL10 | <0.001 | <0.001 | <0.001 | 0.074 |
| CXCL11 | <0.001 | <0.001 | 0.010 | <0.001 |
| GBP1 | <0.001 | <0.001 | 0.001 | <0.001 |
| IDO1 | <0.001 | <0.001 | <0.001 | 0.001 |
| IFNB1 | 0.003 | 1.000 | 1.000 | 1.000 |
| IL27 | <0.001 | <0.001 | 0.013 | NA |
| IL-29 | 0.041 | NA | 1.00 | NA |
| TGFBR2 | <0.001 | 0.014 | 0.009 | 0.433 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kremenovic, M.; Rombini, N.; Chan, A.A.; Gruber, T.; Bäriswyl, L.; Lee, D.J.; Schenk, M. Characterization of a Myeloid Activation Signature That Correlates with Survival in Melanoma Patients. Cancers 2020, 12, 1431. https://doi.org/10.3390/cancers12061431
Kremenovic M, Rombini N, Chan AA, Gruber T, Bäriswyl L, Lee DJ, Schenk M. Characterization of a Myeloid Activation Signature That Correlates with Survival in Melanoma Patients. Cancers. 2020; 12(6):1431. https://doi.org/10.3390/cancers12061431
Chicago/Turabian StyleKremenovic, Mirela, Nives Rombini, Alfred A. Chan, Thomas Gruber, Lukas Bäriswyl, Delphine J. Lee, and Mirjam Schenk. 2020. "Characterization of a Myeloid Activation Signature That Correlates with Survival in Melanoma Patients" Cancers 12, no. 6: 1431. https://doi.org/10.3390/cancers12061431
APA StyleKremenovic, M., Rombini, N., Chan, A. A., Gruber, T., Bäriswyl, L., Lee, D. J., & Schenk, M. (2020). Characterization of a Myeloid Activation Signature That Correlates with Survival in Melanoma Patients. Cancers, 12(6), 1431. https://doi.org/10.3390/cancers12061431