Rlip Depletion Suppresses Growth of Breast Cancer

 and
and
Abstract
:1. Introduction
2. Results
2.1. RALBP1 Gene Expression in Human Breast Cancer
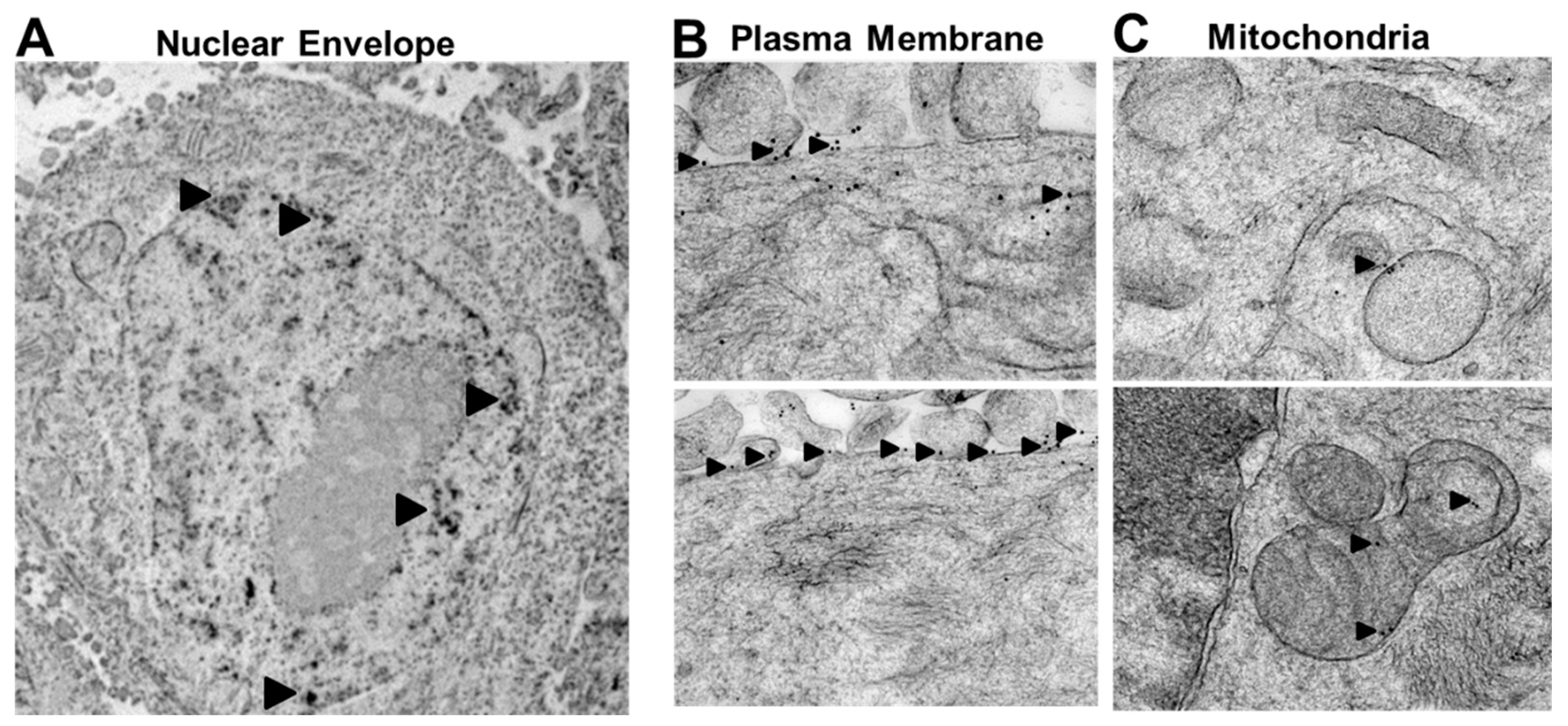
2.2. Rlip Is Localized to Cell Membranes
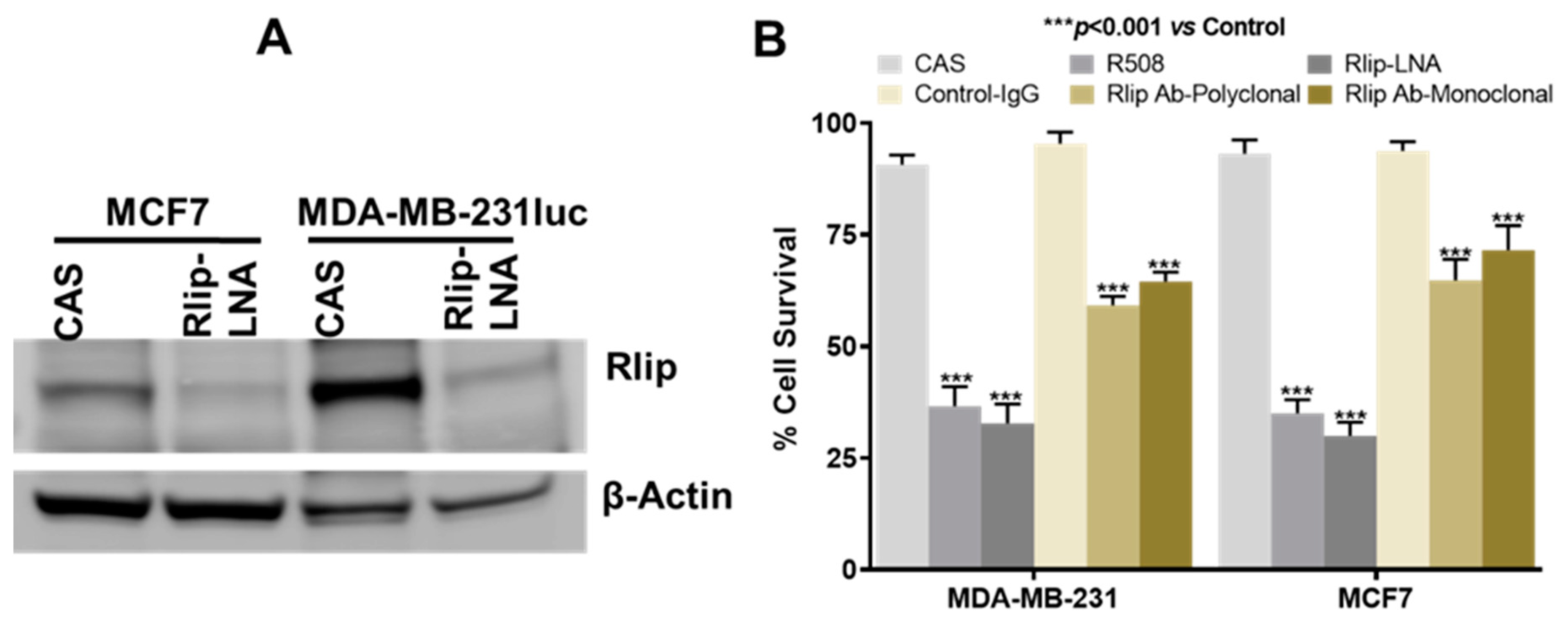
2.3. Anticancer Effects of Rlip Depletion on Breast Cancer Cell Lines In Vitro
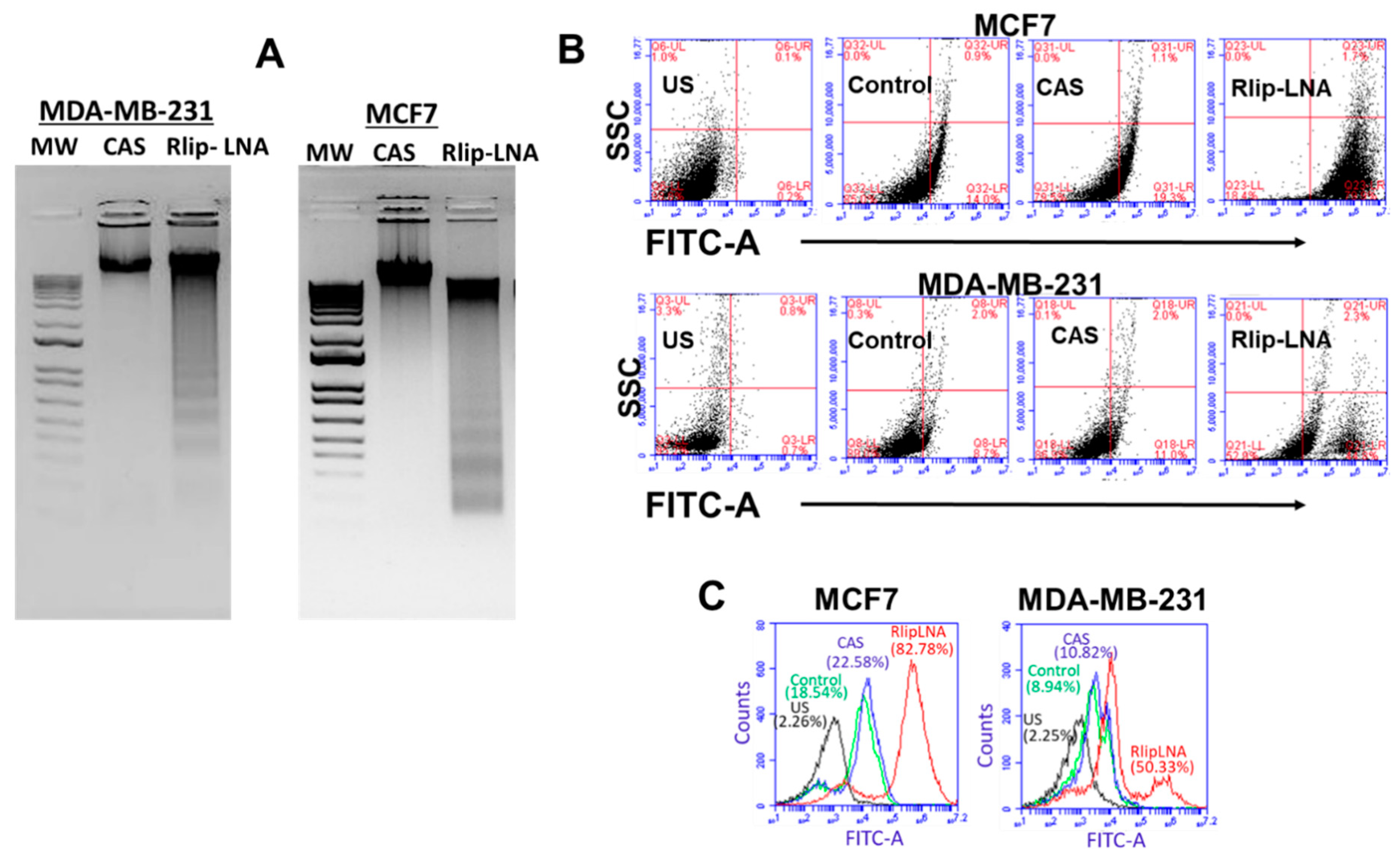
2.4. Rlip-LNA Induces Apoptosis in Breast Cancer Cell Lines In Vitro
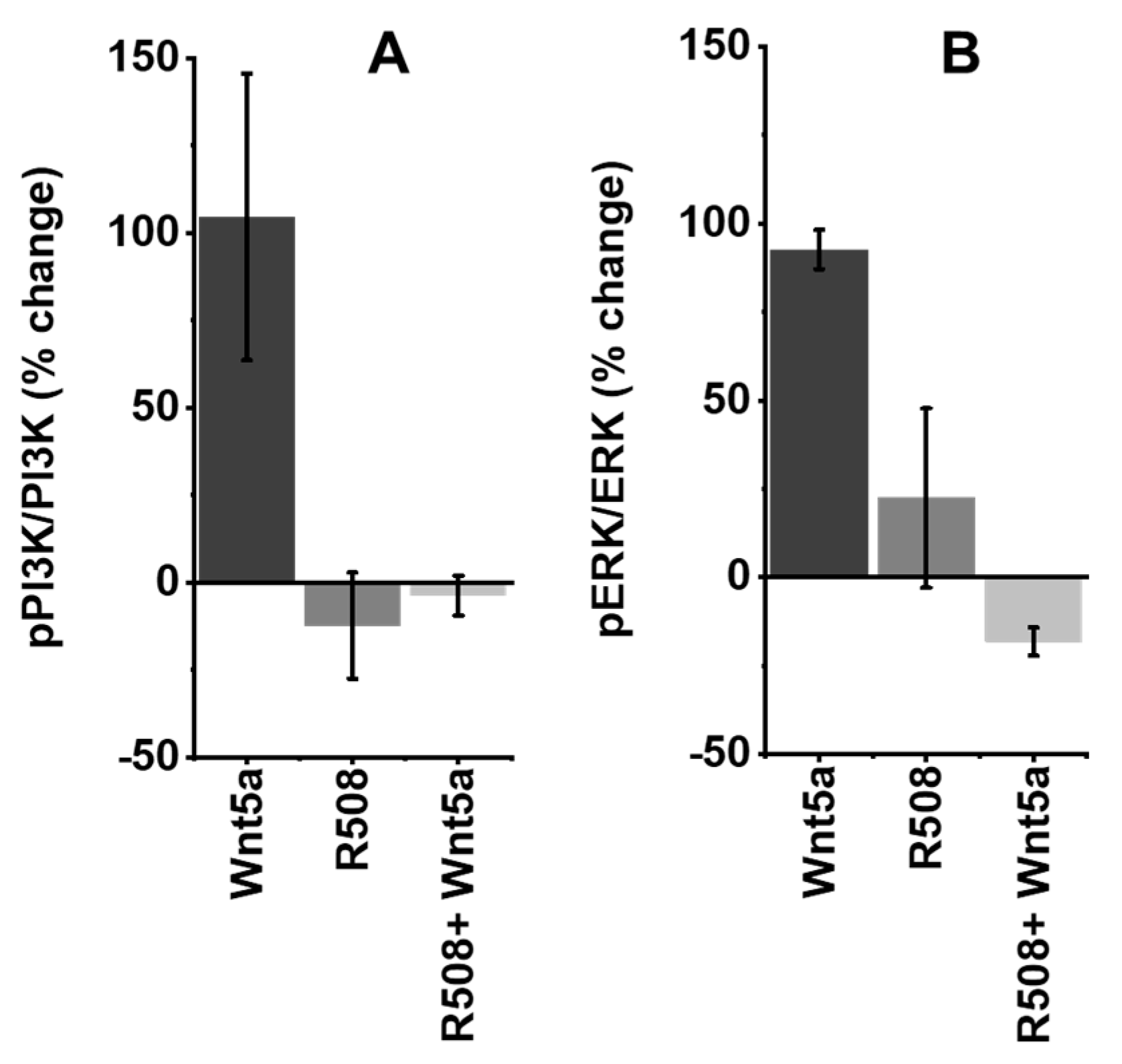
2.5. Role of Rlip in Regulating WNT5A Signaling
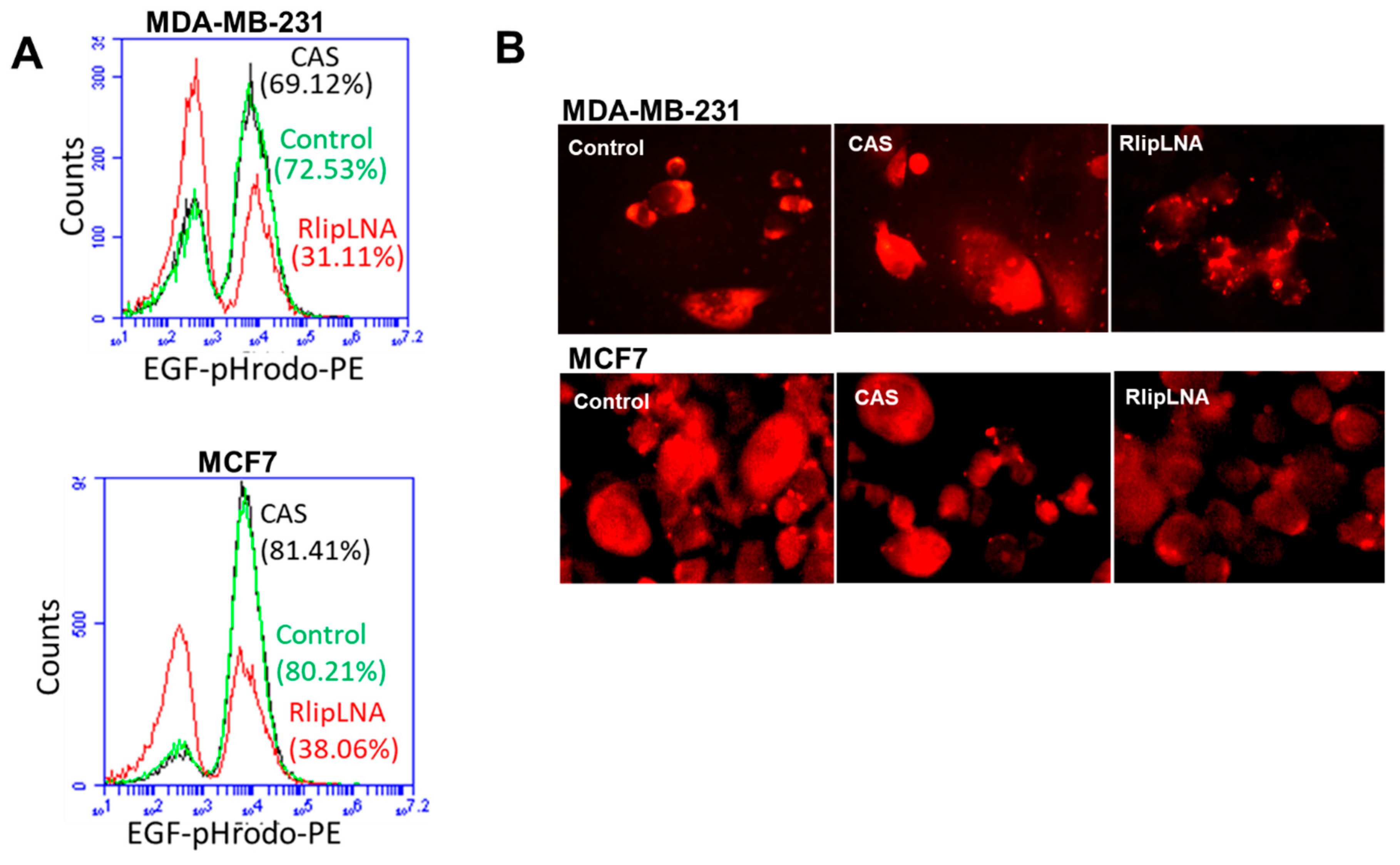
2.6. Rlip-LNA Inhibits Endocytosis in Breast Cancer Cells
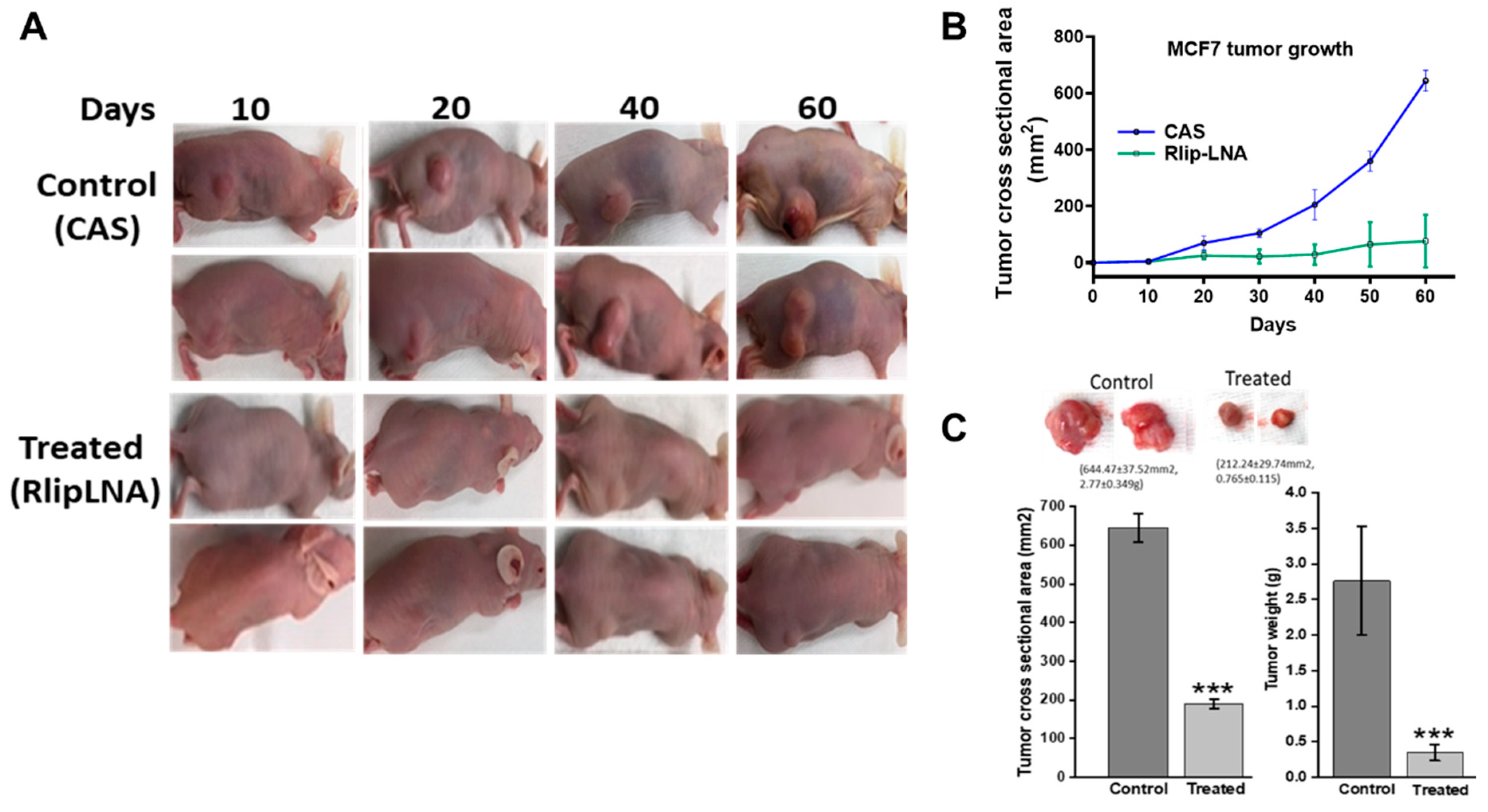
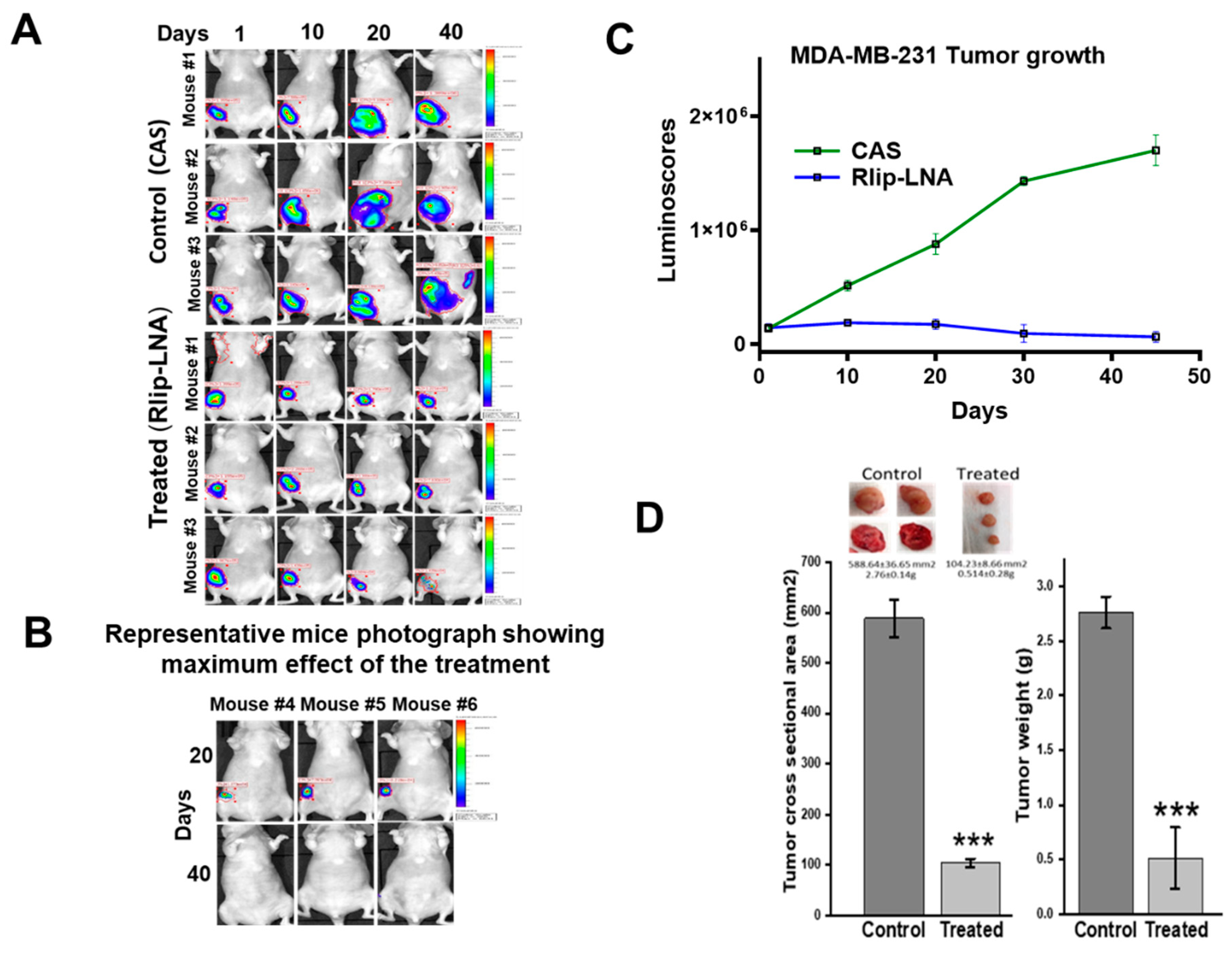
2.7. Anti-Neoplastic Activity of Rlip Targeting in Breast Cancer Xenografts
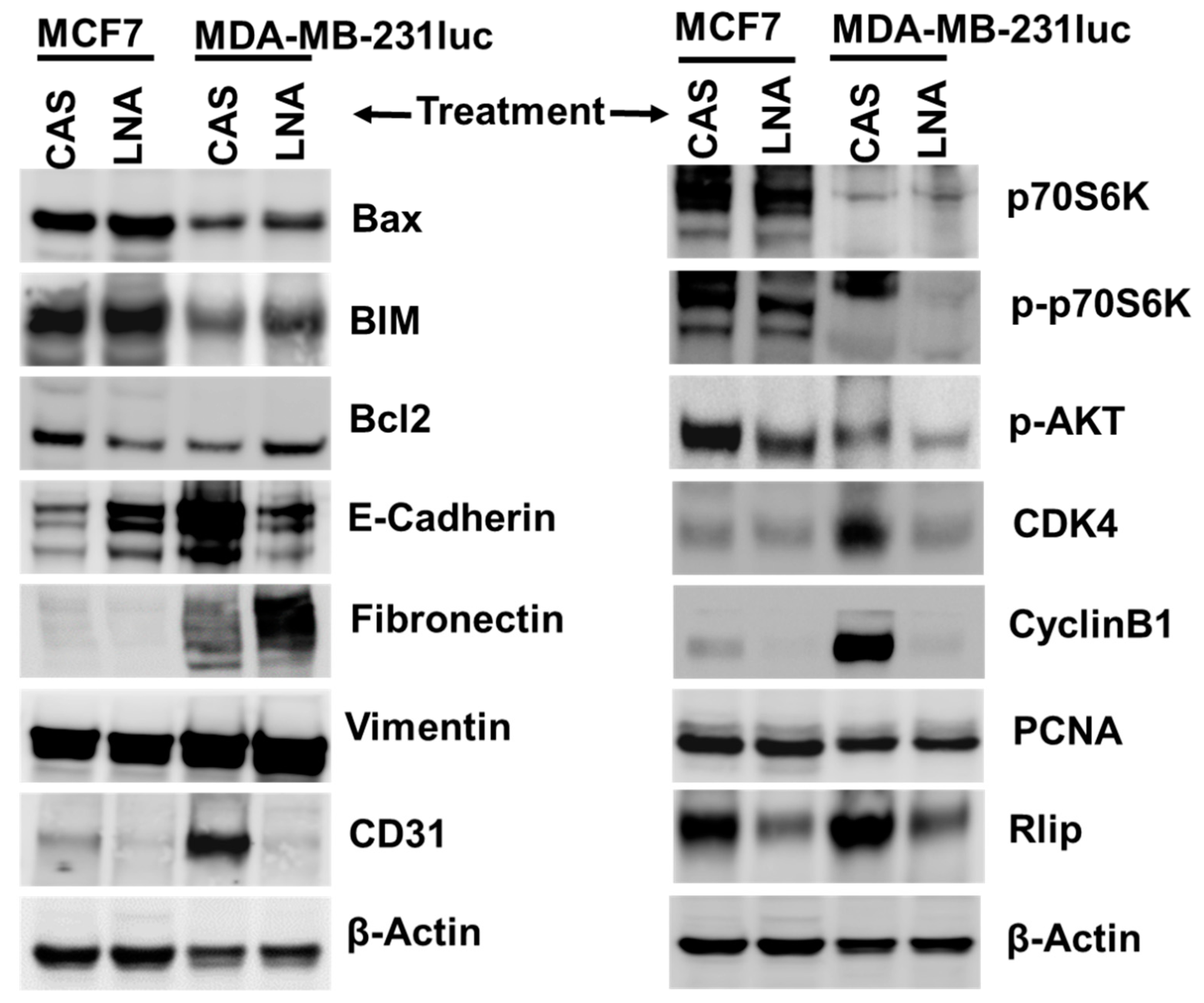
2.8. Targeting Rlip Regulates Cancer Signaling in MCF7 and MDA-MB-231 Xenografts
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cultures
4.3. Mouse Studies
4.4. Ethics Statement
4.5. R508 Antisense and Locked Nucleic Acid (LNA) Oligonucleotides of Rlip
4.6. Cell Proliferation and Cytotoxicity Assay
4.7. Effect of Rlip Depletion on EGF Binding and Internalization Immunofluorescence
4.8. Flow Cytometric Analysis of EGF Endocytosis
4.9. TUNEL Assay
4.10. Determination of Apoptosis by DNA Laddering
4.11. Subcellular Localization of Rlip by Electron Microscopy
4.12. Tumor Xenografts, Treatments, and Imaging
4.13. Assessment of Angiogenesis, Proliferation, and Apoptotic Signaling
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Fedewa, S.A.; Goding Sauer, A.; Kramer, J.L.; Smith, R.A.; Jemal, A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J. Clin. 2016, 66, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. SEER Cancer Statistics Review 1975–2014; National Cancer Institute: Bethesda, MD, USA, 2017. [Google Scholar]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in adjuvant chemotherapy for breast cancer: An overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeichner, S.B.; Terawaki, H.; Gogineni, K. A Review of Systemic Treatment in Metastatic Triple-Negative Breast Cancer. Breast Cancer (Auckl) 2016, 10, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, Y.C.; Sharma, R.; Yadav, S.; Dwivedi, S.; Sharma, A.; Awasthi, S. The non-ABC drug transporter RLIP76 (RALBP-1) plays a major role in the mechanisms of drug resistance. Curr. Drug Metab. 2007, 8, 315–323. [Google Scholar] [CrossRef]
- Giai, M.; Biglia, N.; Sismondi, P. Chemoresistance in breast tumors. Eur. J. Gynaecol. Oncol. 1991, 12, 359–373. [Google Scholar]
- Singhal, S.S.; Singhal, J.; Nair, M.P.; Lacko, A.G.; Awasthi, Y.C.; Awasthi, S. Doxorubicin transport by RALBP1 and ABCG2 in lung and breast cancer. Int. J. Oncol. 2007, 30, 717–725. [Google Scholar] [CrossRef]
- Stuckler, D.; Singhal, J.; Singhal, S.S.; Yadav, S.; Awasthi, Y.C.; Awasthi, S. RLIP76 transports vinorelbine and mediates drug resistance in non-small cell lung cancer. Cancer Res. 2005, 65, 991–998. [Google Scholar]
- Yadav, S.; Zajac, E.; Singhal, S.S.; Awasthi, S. Linking stress-signaling, glutathione metabolism, signaling pathways and xenobiotic transporters. Cancer Metastasis Rev. 2007, 26, 59–69. [Google Scholar] [CrossRef]
- Green, A.R.; Aleskandarany, M.A.; Agarwal, D.; Elsheikh, S.; Nolan, C.C.; Diez-Rodriguez, M.; Macmillan, R.D.; Ball, G.R.; Caldas, C.; Madhusudan, S.; et al. MYC functions are specific in biological subtypes of breast cancer and confers resistance to endocrine therapy in luminal tumours. Br. J. Cancer 2016, 114, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, N.; Leyland-Jones, B.; De, P. MYC-xing it up with PIK3CA mutation and resistance to PI3K inhibitors: Summit of two giants in breast cancers. Am. J. Cancer Res. 2015, 5, 1–19. [Google Scholar] [PubMed]
- Awasthi, S.; Singhal, S.S.; Yadav, S.; Singhal, J.; Drake, K.; Nadkar, A.; Zajac, E.; Wickramarachchi, D.; Rowe, N.; Yacoub, A.; et al. RLIP76 is a major determinant of radiation sensitivity. Cancer Res. 2005, 65, 6022–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat Res. 2016, 186, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Singhal, J.; Singhal, S.S.; Yadav, S.; Suzuki, S.; Warnke, M.M.; Yacoub, A.; Dent, P.; Bae, S.; Sharma, R.; Awasthi, Y.C.; et al. RLIP76 in defense of radiation poisoning. Int. J. Radiat Oncol. Biol. Phys. 2008, 72, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.E. Metastatic Breast Cancer: The Treatment Challenge. Clin. Breast Cancer 2008, 8, 224–233. [Google Scholar] [CrossRef]
- The, L. Breast cancer targeted therapy: Successes and challenges. Lancet 2017, 389, 2350. [Google Scholar]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007, 608, 1–22. [Google Scholar]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Mohammed, M.K.; Shao, C.; Wang, J.; Wei, Q.; Wang, X.; Collier, Z.; Tang, S.; Liu, H.; Zhang, F.; Huang, J.; et al. Wnt/beta-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis. 2016, 3, 11–40. [Google Scholar] [CrossRef] [Green Version]
- Sakaki-Yumoto, M.; Katsuno, Y.; Derynck, R. TGF-beta family signaling in stem cells. Biochim. Biophys. Acta 2013, 1830, 2280–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, Y.C.; Sharma, R.; Singhal, S.S. Human glutathione S-transferases. Int. J. Biochem. 1994, 26, 295–308. [Google Scholar] [CrossRef]
- Jardim, B.V.; Moschetta, M.G.; Leonel, C.; Gelaleti, G.B.; Regiani, V.R.; Ferreira, L.C.; Lopes, J.R.; Zuccari, D.A. Glutathione and glutathione peroxidase expression in breast cancer: An immunohistochemical and molecular study. Oncol. Rep. 2013, 30, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasabova, Z.; Tilandyova, P.; Kajo, K.; Zubor, P.; Burjanivova, T.; Danko, J.; Plank, L. Hypermethylation of the GSTP1 promoter region in breast cancer is associated with prognostic clinicopathological parameters. Neoplasma 2010, 57, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Sharma, R.; Singhal, S.S.; Zimniak, P.; Awasthi, Y.C. RLIP76, a novel transporter catalyzing ATP-dependent efflux of xenobiotics. Drug Metab. Dispos. 2002, 30, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Singhal, J.; Yadav, S.; Sahu, M.; Awasthi, Y.C.; Awasthi, S. RLIP76: A target for kidney cancer therapy. Cancer Res. 2009, 69, 4244–4251. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Sharma, R.; Awasthi, Y.C.; Belli, J.A.; Frenkel, E.P. The relationship of doxorubicin binding to membrane lipids with drug resistance. Cancer Lett. 1992, 63, 109–116. [Google Scholar] [CrossRef]
- Drake, K.J.; Singhal, J.; Yadav, S.; Nadkar, A.; Pungaliya, C.; Singhal, S.S.; Awasthi, S. RALBP1/RLIP76 mediates multidrug resistance. Int. J. Oncol. 2007, 30, 139–144. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Cheng, J.; Zimniak, P.; Awasthi, Y.C. Role of RLIP76 in lung cancer doxorubicin resistance: II. Doxorubicin transport in lung cancer by RLIP76. Int. J. Oncol. 2003, 22, 713–720. [Google Scholar] [CrossRef]
- Cantor, S.B.; Urano, T.; Feig, L.A. Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol. Cell Biol. 1995, 15, 4578–4584. [Google Scholar] [CrossRef] [Green Version]
- Jullien-Flores, V.; Dorseuil, O.; Romero, F.; Letourneur, F.; Saragosti, S.; Berger, R.; Tavitian, A.; Gacon, G.; Camonis, J.H. Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem. 1995, 270, 22473–22477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinaka, K.; Koyama, S.; Nakashima, S.; Hinoi, T.; Okawa, K.; Iwamatsu, A.; Kikuchi, A. Epsin binds to the EH domain of POB1 and regulates receptor-mediated endocytosis. Oncogene 1999, 18, 5915–5922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Wickramarachchi, D.; Yadav, S.; Singhal, J.; Leake, K.; Vatsyayan, R.; Chaudhary, P.; Lelsani, P.; Suzuki, S.; Yang, S.; et al. Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol. Cancer Ther. 2011, 10, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Awasthi, Y.C.; Yang, Y.; Sharma, A.; Singhal, S.S.; Awasthi, S. Energy dependent transport of xenobiotics and its relevance to multidrug resistance. Curr. Cancer Drug Targets 2003, 3, 89–107. [Google Scholar] [CrossRef]
- Feng, Q.; Gao, N. Keeping Wnt signalosome in check by vesicular traffic. J. Cell Physiol. 2015, 230, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Leake, K.; Singhal, J.; Nagaprashantha, L.; Awasthi, S.; Singhal, S.S. RLIP76 Regulates PI3K/Akt Signaling and Chemo-Radiotherapy Resistance in Pancreatic Cancer. PLoS ONE 2012, 7, e34582. [Google Scholar] [CrossRef] [Green Version]
- Singhal, J.; Yadav, S.; Nagaprashantha, L.D.; Vatsyayan, R.; Singhal, S.S.; Awasthi, S. Targeting p53-null neuroblastomas through RLIP76. Cancer Prev. Res. (Phila) 2011, 4, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Awasthi, Y.C.; Awasthi, S. Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res. 2006, 66, 2354–2360. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Roth, C.; Leake, K.; Singhal, J.; Yadav, S.; Awasthi, S. Regression of prostate cancer xenografts by RLIP76 depletion. Biochem. Pharmacol. 2009, 77, 1074–1083. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Singhal, J.; Yadav, S.; Dwivedi, S.; Boor, P.J.; Awasthi, Y.C.; Awasthi, S. Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76 (Ral-binding protein 1). Cancer Res. 2007, 67, 4382–4389. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Tompkins, J.; Singhal, J.; Riggs, A.D.; Yadav, S.; Wu, X.; Singh, S.; Warden, C.; Liu, Z.; Wang, J.; et al. Rlip depletion prevents spontaneous neoplasia in TP53 null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 3918–3923. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Singhal, S.S.; Singhal, J.; Wickramarachchi, D.; Knutson, E.; Albrecht, T.B.; Awasthi, Y.C.; Awasthi, S. Identification of membrane-anchoring domains of RLIP76 using deletion mutant analyses. Biochemistry 2004, 43, 16243–16253. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Yang, Y.; Zimniak, P.; Awasthi, Y.C. Role of RLIP76 in lung cancer doxorubicin resistance: III. Anti-RLIP76 antibodies trigger apoptosis in lung cancer cells and synergistically increase doxorubicin cytotoxicity. Int. J. Oncol. 2003, 22, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillatre, J.; Delacour, D.; Van Hove, L.; Bagarre, T.; Houssin, N.; Soulika, M.; Veitia, R.A.; Moreau, J. Dynamics of the subcellular localization of RalBP1/RLIP through the cell cycle: The role of targeting signals and of protein-protein interactions. FASEB J. 2012, 26, 2164–2174. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, A.; Yang, Y.; Awasthi, S.; Singhal, S.S.; Zimniak, P.; Awasthi, Y.C. Functional reconstitution of Ral-binding GTPase activating protein, RLIP76, in proteoliposomes catalyzing ATP-dependent transport of glutathione conjugate of 4-hydroxynonenal. Acta Biochim. Pol. 2002, 49, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Figarola, J.; Singhal, J.; Reddy, M.A.; Liu, X.; Berz, D.; Natarajan, R.; Awasthi, S. RLIP76 protein knockdown attenuates obesity due to a high-fat diet. J. Biol. Chem. 2013, 288, 23394–23406. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Yadav, S.; Singhal, J.; Zajac, E.; Awasthi, Y.C.; Awasthi, S. Depletion of RLIP76 sensitizes lung cancer cells to doxorubicin. Biochem. Pharmacol. 2005, 70, 481–488. [Google Scholar] [CrossRef]
- Singhal, J.; Chikara, S.; Horne, D.; Salgia, R.; Awasthi, S.; Singhal, S.S. 2’-Hydroxyflavanone inhibits in vitro and in vivo growth of breast cancer cells by targeting RLIP76. Mol. Carcinog. 2018, 57, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Chikara, S.; Horne, D.; Salgia, R.; Awasthi, S.; Singhal, S.S. RLIP inhibition suppresses breast-to-lung metastasis. Cancer Lett. 2019, 447, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Horne, D.; Singhal, J.; Vonderfecht, S.; Salgia, R.; Awasthi, S. Synergistic efficacy of RLIP inhibition and 2′-hydroxyflavanone against DMBA-induced mammary carcinogenesis in SENCAR mice. Mol Carcinog. 2019, 58, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Rennoll, S.; Yochum, G. Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290–300. [Google Scholar] [CrossRef]
- Mettlen, M.; Chen, P.-H.; Srinivasan, S.; Danuser, G.; Schmid, S.L. Regulation of Clathrin-Mediated Endocytosis. Annu. Rev. Biochem. 2018, 87, 871–896. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Bose, C.; Singh, S.P.; Igid, H.; Green, W.C.; Singhal, S.S.; Lee, J.; Palade, P.T.; Rajan, A.; Ball, S.; Tonk, V.; et al. Topical 2′-Hydroxyflavanone for Cutaneous Melanoma. Cancers 2019, 11, 1556. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Nagaprashantha, L.; Li, H.; Yuan, Y.-C.; Liu, Z.; Berz, D.; Igid, H.; Green, W.C.; et al. Anticancer activity of 2-hydroxyflavanone towards lung cancer. Oncotarget 2018, 9, 36202–36219. [Google Scholar] [CrossRef]
- Singhal, S.S.; Yadav, S.; Drake, K.; Singhal, J.; Awasthi, S. Hsf-1 and POB1 induce drug sensitivity and apoptosis by inhibiting Ralbp1. J. Biol. Chem. 2008, 283, 19714–19729. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Wurtzel, J.G.; Singhal, S.S.; Awasthi, S.; Goldfinger, L.E. RALBP1/RLIP76 depletion in mice suppresses tumor growth by inhibiting tumor neovascularization. Cancer Res. 2012, 72, 5165–5173. [Google Scholar] [CrossRef] [Green Version]
- Mollberg, N.M.; Steinert, G.; Aigner, M.; Hamm, A.; Lin, F.-J.; Elbers, H.; Reissfelder, C.; Weitz, J.; Buchler, M.W.; Koch, M. Overexpression of RalBP1 in colorectal cancer is an independent predictor of poor survival and early tumor relapse. Cancer Biol. Ther. 2012, 13, 694–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Singhal, S.S.; Yadav, S.; Singhal, J.; Vatsyayan, R.; Zajac, E.; Luchowski, R.; Borvak, J.; Gryczynski, K.; Awasthi, Y.C. A central role of RLIP76 in regulation of glycemic control. Diabetes 2010, 59, 714–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, J.; Nagaprashantha, L.; Vatsyayan, R.; Awasthi, S.; Singhal, S.S. RLIP76, a glutathione-conjugate transporter, plays a major role in the pathogenesis of metabolic syndrome. PLoS ONE 2011, 6, e24688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monks, T.J.; Lau, S.S. Glutathione, gamma-glutamyl transpeptidase, and the mercapturic acid pathway as modulators of 2-bromohydroquinone oxidation. Toxicol. Appl. Pharmacol. 1990, 103, 557–563. [Google Scholar] [CrossRef]
- Hinchman, C.A.; Ballatori, N. Glutathione conjugation and conversion to mercapturic acids can occur as an intrahepatic process. J. Toxicol. Environ. Health 1994, 41, 387–409. [Google Scholar] [CrossRef]
- Awasthi, S.; Srivastava, S.K.; Ahmad, F.; Ahmad, H.; Ansari, G.A. Interactions of glutathione S-transferase-pi with ethacrynic acid and its glutathione conjugate. Biochim. Biophys. Acta 1993, 1164, 173–178. [Google Scholar] [CrossRef]
- Rousar, T.; Parik, P.; Kucera, O.; Bartos, M.; Cervinkova, Z. Glutathione reductase is inhibited by acetaminophen-glutathione conjugate in vitro. Physiol. Res. 2010, 59, 225–232. [Google Scholar]
- Lehnert, S.; Greene, D.; Batist, G. Radiation response of drug-resistant variants of a human breast cancer cell line: The effect of glutathione depletion. Radiat. Res. 1990, 124, 208–215. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Pikula, S.; Piper, J.T.; Srivastava, S.K.; Torman, R.T.; Bandorowicz-Pikula, J.; Lin, J.T.; Singh, S.V.; Zimniak, P.; et al. ATP-Dependent human erythrocyte glutathione-conjugate transporter. II. Functional reconstitution of transport activity. Biochemistry 1998, 37, 5239–5248. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Srivastava, S.K.; Torman, R.T.; Zimniak, P.; Bandorowicz-Pikula, J.; Singh, S.V.; Piper, J.T.; Awasthi, Y.C.; Pikula, S. ATP-Dependent human erythrocyte glutathione-conjugate transporter. I. Purification, photoaffinity labeling, and kinetic characteristics of ATPase activity. Biochemistry 1998, 37, 5231–5238. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Srivastava, S.K.; Zimniak, P.; Bajpai, K.K.; Saxena, M.; Sharma, R.; Ziller, S.A., 3rd; Frenkel, E.P.; Singh, S.V.; et al. Adenosine triphosphate-dependent transport of doxorubicin, daunomycin, and vinblastine in human tissues by a mechanism distinct from the P-glycoprotein. J. Clin. Invest. 1994, 93, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbar Saadat, Y.; Saeidi, N.; Zununi Vahed, S.; Barzegari, A.; Barar, J. An update to DNA ladder assay for apoptosis detection. Bioimpacts 2015, 5, 25–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplified Genes | Locus | Rlip-Unaltered | Rlip-High | Rlip-Low | Function |
| MYC | 8q24.21 | 25.49 | 53.42 | 25.69 | proto-oncogene |
| RAD21 | 8q24.11 | 24.85 | 50.68 | 25.23 | sister chromatid cohesion in mitotic cells |
| EXT1 | 8q24.11 | 24.39 | 48.63 | 24.31 | chain elongation step of heparan sulfate |
| NDRG1 | 8q24.22 | 23.15 | 48.63 | 22.94 | stress responses, hormone responses, cell growth, and differentiation |
| RSPO2 | 8q23.1 | 23.15 | 43.15 | 23.85 | signaling receptor binding and G protein-coupled receptor binding |
| Deleted Genes | Locus | Rlip-Unaltered | Rlip-High | Rlip-Low | Function |
| CDKN2A | 9p21.3 | 2.44 | 2.05 | 4.59 | cell cycle G1 control |
| CDKN2B | 9p21.3 | 2.44 | 2.05 | 4.59 | cell cycle G1 progression |
| MAP2K4 | 17p12 | 2.12 | 0.68 | 2.75 | transferase activity and protein tyrosine kinase activity |
| PTEN | 10q23.31 | 2.02 | 2.74 | 1.83 | multi-functional tumor suppressor |
| MTAP | 9p21.3 | 2.3 | 2.05 | 4.59 | phosphorylase activity and S-methyl-5-thioadenosine phosphorylase activity |
| Mutated Genes | Locus | Rlip-Unaltered | Rlip-High | Rlip-Low | Function |
| PIK3CA | 3q26.32 | 42.52 | 16.55 | 45.07 | transferring phosphorus-containing groups and protein serine/threonine kinase activity |
| TP53 | 17p13.1 | 34.66 | 48.97 | 45.07 | DNA-binding transcription factor activity and protein heterodimerization activity |
| MUC16 | 19p13.2 | 16.81 | 15.86 | 22.54 | cell surface glycosylation |
| AHNAK2 | 14q32.33 | 16.71 | 11.72 | 19.72 | calcium signaling by associating with calcium channel proteins |
| SYNE1 | 6q25.2 | 12.38 | 13.10 | 12.21 | nucleotide binding |
| Prognostic Subsets | Relapse-Free Survival (Months) | Overall Survival (Months) | ||||||
|---|---|---|---|---|---|---|---|---|
| Patient Number | Rlip-Low | Rlip-High | -logp | Patient Number | Rlip-Low | Rlip-High | -logp | |
| All | 3951 | 61 | 34 | 10.5 | 1402 | 124 | 82 | 1.4 |
| Luminal androgen receptor | 203 | 30 | 12 | 8.7 | 83 | 52 | 16 | 1.9 |
| Her2 + | 252 | 32 | 12 | 5.4 | 129 | 72 | 35 | 1.3 |
| Luminal A | 1933 | 93 | 51 | 4.9 | 611 | 151 | 99 | 1.1 |
| Basal 1 | 171 | 39 | 16 | 4.0 | 58 | n.a. | n.a. | 1.8 |
| Luminal B | 1149 | 49 | 35 | 3.9 | 433 | 97 | 70 | 1.1 |
| LN + | 1133 | 171 | 122 | 2.6 | 313 | 56 | 36 | 1.5 |
| Any chemotherapy | 798 | 55 | 36 | 2.6 | 300 | n.a. | n.a. | 1.5 |
| Grade 3 | 903 | 37 | 28 | 2.1 | 503 | 64 | 45 | 1.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bose, C.; Yadav, S.; Singhal, S.S.; Singhal, J.; Hindle, A.; Lee, J.; Cheedella, N.K.S.; Rehman, S.; Layeequr Rahman, R.; Jones, C.; et al. Rlip Depletion Suppresses Growth of Breast Cancer. Cancers 2020, 12, 1446. https://doi.org/10.3390/cancers12061446
Bose C, Yadav S, Singhal SS, Singhal J, Hindle A, Lee J, Cheedella NKS, Rehman S, Layeequr Rahman R, Jones C, et al. Rlip Depletion Suppresses Growth of Breast Cancer. Cancers. 2020; 12(6):1446. https://doi.org/10.3390/cancers12061446
Chicago/Turabian StyleBose, Chhanda, Sushma Yadav, Sharad S. Singhal, Jyotsana Singhal, Ashly Hindle, Jihyun Lee, Naga K. S. Cheedella, Shabnam Rehman, Rakhshanda Layeequr Rahman, Catherine Jones, and et al. 2020. "Rlip Depletion Suppresses Growth of Breast Cancer" Cancers 12, no. 6: 1446. https://doi.org/10.3390/cancers12061446
APA StyleBose, C., Yadav, S., Singhal, S. S., Singhal, J., Hindle, A., Lee, J., Cheedella, N. K. S., Rehman, S., Layeequr Rahman, R., Jones, C., Darden, M., Palade, P. T., Berz, D., Singh, S. P., & Awasthi, S. (2020). Rlip Depletion Suppresses Growth of Breast Cancer. Cancers, 12(6), 1446. https://doi.org/10.3390/cancers12061446